What is your diagnosis?
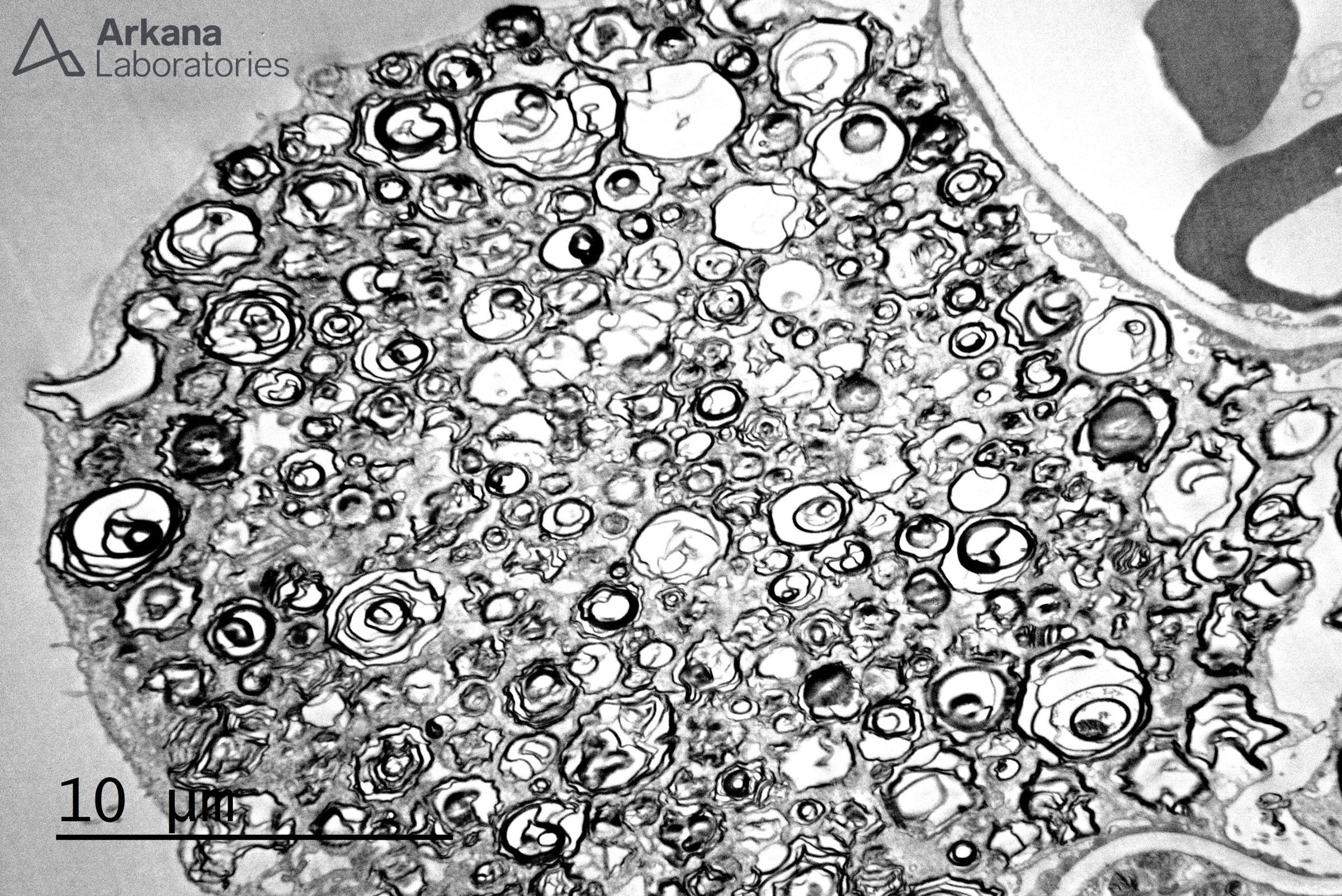
The electron microscopic image depicts a podocyte with numerous myeloid bodies, myelinosomes, or “zebra” bodies as they are also known. This finding, taken in concert with the patient’s clinical history was compatible with Fabry’s disease. By light microscopy, a characteristic finding is the lacy, or foamy cytoplasm of the podocytes due to the lipid inclusions composed of globotriaosylceramide. Additionally, toluidine blue sections can be extremely helpful in identifying these inclusions.
Fabry’s disease is classified as a lysosomal storage disease caused by a deficiency in the alpha-galactosidase, an enzyme leading to the accumulation of globotriaosylceramide (GL3) not only in the kidney but other organs such as the heart, sweat glands, and nerves. Accumulation of GL3 occurs in endothelial cells, smooth muscle cells, podocytes, distal tubular cells, cardiac myocytes, and dorsal root ganglia, among others. While Fabry’s disease is caused by an X-linked mutation and typically affects males, heterozygous females can also have variable manifestations of the disease. Of note, other known causes of lipid accumulation in podocytes include chloroquine or hydroxychloroquine therapy; however, this is typically a focal finding in these cases.
The podocyte changes present in this biopsy are consistent with Fabry disease. Although unlikely, the differential diagnosis includes iatrogenic phospholipidosis secondary to drugs such as chloroquine, amiodarone, aminoglycoside antibiotics, antidepressants, and anti-cholesterol medications among others (see reference below). Correlation with other diagnostic tests such as alpha galactosidase activity and/or genetic analysis is required for definitive diagnosis.
References:
- Woywodt A, Hellweg S, et al. A wild zebra chase. Nephrol Dial Transplant 2007;22(10):3074-7.
- Bracamonte ER, Kowalewska J, et al. Iatrogenic phospholipidosis mimicking Fabry disease. Am J Kidney Dis 2006; 48:844-850.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.