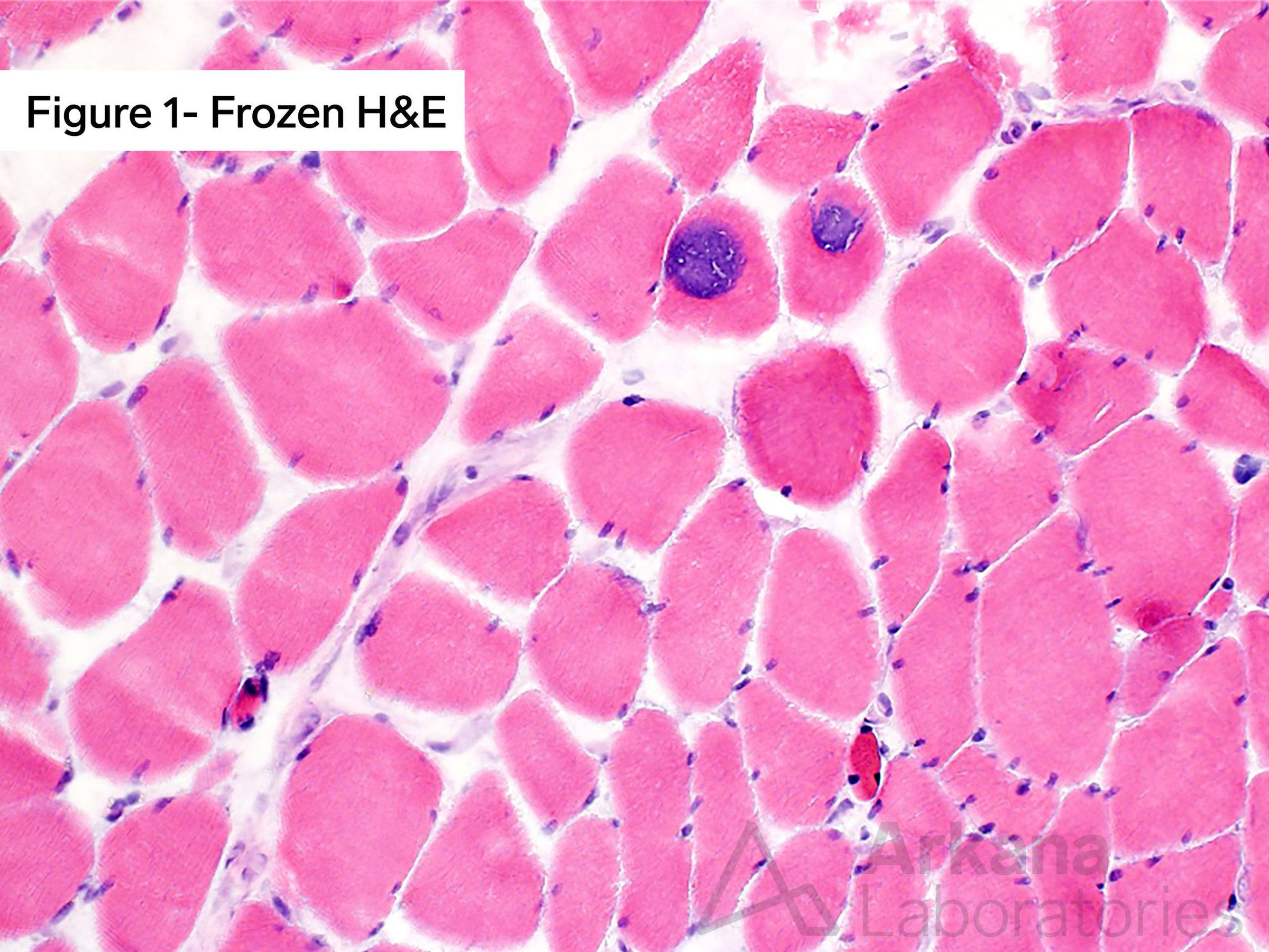
Considering the clinical vignette from the prior week (elderly male with proximal muscle weakness). Staining the skeletal muscle biopsy with Hematoxylin and Eosin (H&E) in combination with frozen sections evaluated with Modified Gomori Trichrome (MGT) and oxidative stains, as well as electron microscopy, demonstrated findings diagnostic of myopathy with tubular aggregates (TAM). See the images. Mutation in which of the following genes is associated with myopathy with tubular aggregates?
Frozen tissue section of skeletal muscle with Tubular Aggregate Myopathy (TAM) demonstrating two adjacent muscle fibers with round to ovoid eccentric and subsarcolemmal inclusion-type material with dense granular basophilic (blue) staining character (A) with hematoxylin and eosin (H&E) staining. Ultrastructural examination by electron microscopy from the case shows scattered muscle fibers with variably sized sarcoplasmic collections of tubular aggregates (B). (Original magnifications: A. Frozen hematoxylin and eosin (H&E) stain, 100x; B. Electron microscopy, 8000x).

Answer: STIM1 and ORAI1.
Myopathy cases with frequent aggregates on skeletal muscle biopsy are classified as “tubular aggregate myopathy (TAM)”. Recently, mutations in two genes, STIM1 (TAM1) and ORAI1 (TAM2), have been identified in familial cases of TAM, but these cases typically present in younger patients. Four phenotypes have been described: (1) TAM with isolated progressive muscle weakness, (2) TAM with exercise-induced cramps, pain, and stiffness; (3) TAM with myasthenic features, and (4) TAM with gyrate atrophy of the choroids and retina. Other less commonly reported clinical manifestations in TAM patients include malignant hyperthermia and acute respiratory failure.
Of note, tubular aggregates are not specific as to etiology as they may be seen in a variety of both acquired and inherited conditions. The acquired conditions include alcoholic myopathy, periodic paralysis (hypo-, hyper-, normokalemic, and thyrotoxic), neuropathy, motor neuron disease, inflammatory myopathy, and acromegaly.
Tubular aggregates have also been seen in the setting of phosphoglycerate mutase deficiency (PGAM1 gene), limb-girdle myasthenia associated with GFPT1 mutations, and myotonic dystrophy.
Why were the other answers wrong?
Alterations in DMD are associated with dystrophinopathies, Duchenne muscular dystrophy and Becker muscular dystrophy. Alterations in TTN are associated with titinopathies.
Mutations in CACNA1S and RYR1 are associated with channelopathies, including autosomal dominant hypokalemia periodic paralysis and autosomal dominant/recessive congenital myopathy; and central core disease, autosomal recessive multiminicore disease, and malignant hyperthermia susceptibility (RYR1 and CACNA1S), respectively.
There is significant clinical overlap between Pompe disease (GAA, GSD II) and X-linked Danon disease (LAMP2), and differentiating between these disorders by clinical symptoms alone can be difficult. Muscle biopsy and genetic testing may be helpful.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.