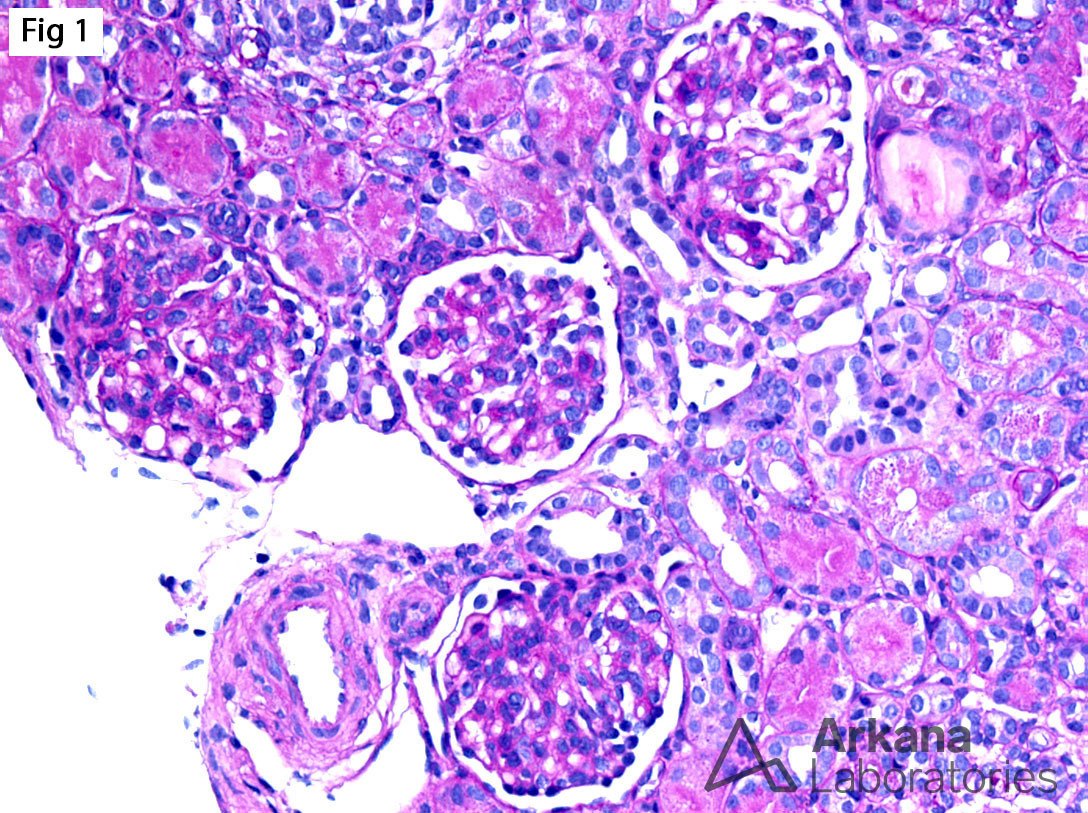
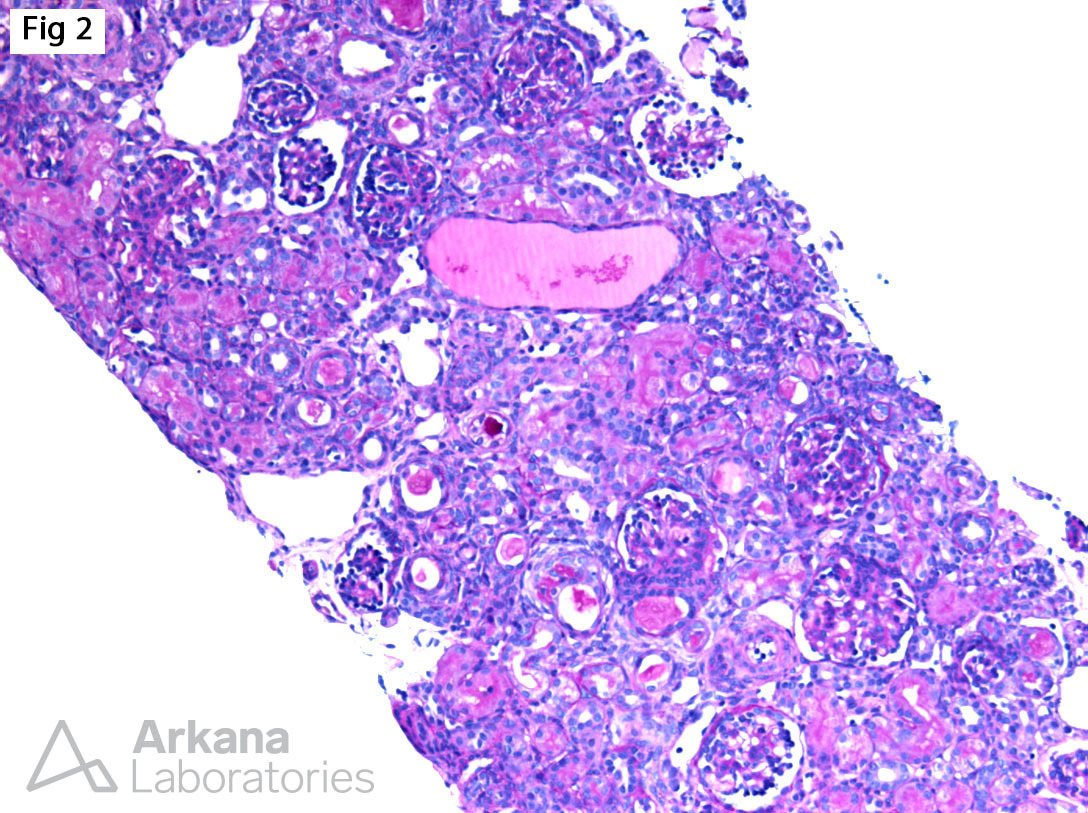
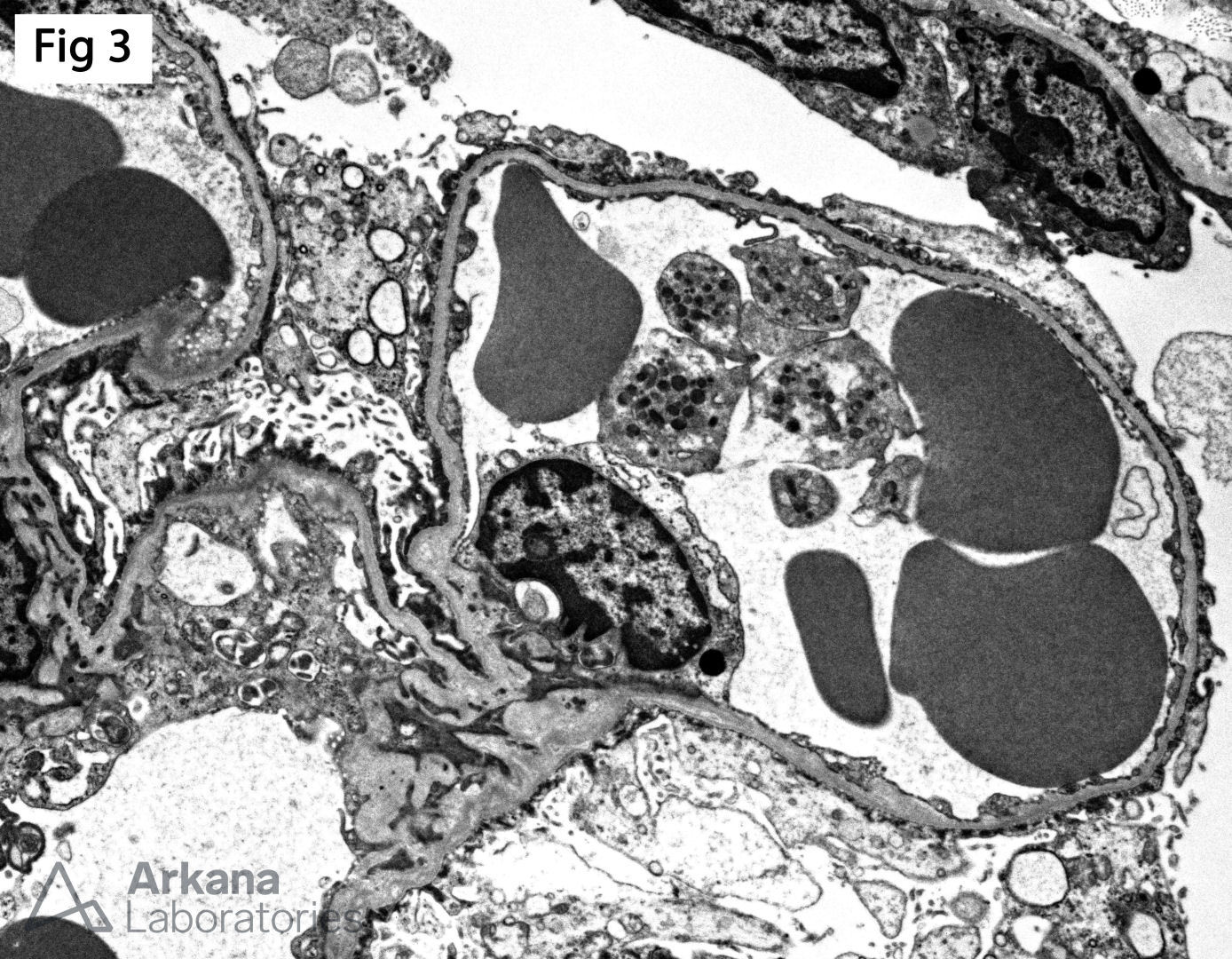
The biopsy from this 5-year-old male with nephrotic syndrome shows immature glomeruli without segmental glomerulosclerosis (Fig 1), associated with focal microcystic tubular dilatation and mild interstitial fibrosis (Fig 2). Furthermore, electron microscopy shows diffuse epithelial foot process effacement with microvillous transformation (Fig 3). In the setting of nephrotic syndrome presenting in utero or in early infancy, the findings are highly suggestive of a congenital nephrotic syndrome of the Finnish type. This entity is caused by a homozygous mutation in NPHS1, which encodes nephrin. Nephrin is a major structural protein of the glomerular filtration slit diaphragm, which links the foot processes of podocytes. Abnormal nephrin results in lack of slit diaphragms and podocyte dysfunction which results in proteinuria. The disease is unresponsive to therapy, and most patients progress to end-stage renal disease by 32 months. On the other hand, patients with heterozygous NPHS1 mutations have an excellent prognosis with normal renal function.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.