
Monday
Type III collagen glomerulopathy was first described by Arakawa in 1979 in Japan. In this first report it was dubbed ‘Idiopathic mesangio-degenerative glomerulopathy.’ Arakawa M. Idiopathic mesangio-degenerative glomerulopathy. Jpn J Nephrol1979; 21: 914–915
Collagen type III glomerulopathy is an ultra-rare kidney disease with approximately 40 cases reported in the literature. Reports from Japan are most common but reports are widespread. https://www.ncbi.nlm.nih.gov/pubmed/26622504
Collagen type III glomerulopathy is caused by the abnormal accumulation of type III collagen within the glomerular mesangium and capillary walls. https://www.ncbi.nlm.nih.gov/pubmed/28532638
Collagen type III glomerulopathy is also sometimes called collagenofibrotic glomerulopathy, especially in the past. However, in recent times, this name is becoming less common.
Tuesday
Collagen type III glomerulopathy is associated with proteinuria and progressive CKD of varying degree. However, many patients progress to ESKD within 10 years. https://www.ncbi.nlm.nih.gov/pubmed/28532638
The etiology and pathogenesis of collagen type III glomerulopathy are unknown, although in a subset of patients (usually pediatric), an AR inheritance pattern has been identified. https://www.ncbi.nlm.nih.gov/pubmed/8398640
The largest case series of collagen type III glomerulopathy in pediatric patients showed a high frequency of superimposed hemolytic uremic syndrome. https://www.ncbi.nlm.nih.gov/pubmed/8398640
In one reported case, inherited factor H deficiency and collagen type III glomerulopathy were both identified in a child. https://www.ncbi.nlm.nih.gov/pubmed/7742208
Wednesday
In adult patients, collagen type III glomerulopathy is typically sporadic, with no identifiable pattern of inheritance. No genetic etiology for the disease has been identified. https://www.ncbi.nlm.nih.gov/pubmed/26413279
Many unusual associations with collagen type III glomerulopathy have been made in case reports ranging from perisinusoidal liver fibrosis to Hodgkin’s lymphoma but these have been absent in larger case series. https://www.ncbi.nlm.nih.gov/pubmed/8450910 https://www.ncbi.nlm.nih.gov/pubmed/21196628
Some believe collagen type III glomerulopathy is a systemic disease as increased serum/urine procollagen type III peptide can be seen. However, this increase can be seen in advanced renal fibrosis and is not specific. https://www.ncbi.nlm.nih.gov/pubmed/9915277
Thursday
Collagen type III glomerulopathy typically has a lobular appearance and double contour formation is often seen.
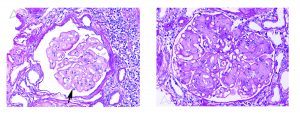
By immunofluorescence and EM, collagen type III glomerulopathy shows no immune complex type deposits. https://www.ncbi.nlm.nih.gov/pubmed/26413279
EM is diagnostic for collagen type III glomerulopathy. Stacked, vaguely curvilinear, fibrillar collagen is seen in the mesangium and capillary walls in routine EM.
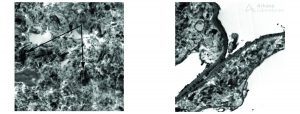
Impressive curvilinear morphology of the fibrils is a useful EM hallmark of collagen type III glomerulopathy and can be visualized when phosphotungstic acid is utilized in EM processing. See figure 4. https://www.ajkd.org/article/S0272-6386(17)30630-3/fulltext
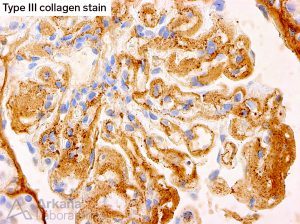
IHC staining for type III collagen is available and useful for diagnostic confirmation in collagen type III glomerulopathy.
Friday
Collagen type III glomerulopathy is an ultra-rare kidney disease of unknown etiology and pathogenesis with only rare cases reported in the literature, the vast majority being case reports. A few non-controlled, small case series exist that shed light on differences in pediatric and adult presentations and specific geographic patient populations.
Type III collagen is typically absent in normal glomeruli, although small amounts have been reported in some advanced glomerular diseases. In patients with collagen type III glomerulopathy, abundant type III collagen is seen within the mesangium and lamina rara interna of the capillary loops and typically occurs in a flocculent mesangial/capillary wall matrix.
Exactly how the type III collagen gets into the mesangium and capillary wall is unknown. Some authors believe this is a systemic disease with circulating procollagen leading to the accumulation of type III collagen. This hypothesis comes from evidence in humans and canines of increased amounts of the serum aminoterminal propeptide of type III procollagen in those with collagen type III glomerulopathy and rare case reports of multi-organ identification of type III collagen deposition. However, the aminoterminal propeptide of type III procollagen is not a specific biomarker and most patients with collagen type III glomerulopathy do not develop symptoms of multi-organ injury in the course of their disease. Conversely, some authors believe that the collagen type III is locally produced in the glomerulus as small amounts of collagen type III can be seen in some advanced glomerular disorders implying the mechanism for production may exist in the injured glomerulus. Also, mesangial cell activation has been identified in at least one report of collagen type III glomerulopathy.
While no genetic etiology has been identified in humans, pediatric forms of the disease can display an autosomal recessive pattern of inheritance. And, a naturally occurring canine equivalent disease displays the same inheritance pattern but without the hypocomplementemia seen in some pediatric patients. Importantly, when diagnosed in adulthood, the disease appears to be sporadic with no inheritance pattern identified and does not typically co-occur with hemolytic uremic syndrome as has also been seen in pediatric patients.
In adult patients, progressive chronic kidney disease usually occurs and some patients progress to end-stage kidney disease while in the pediatric presentation, renal outcomes are generally poor. To date, no controlled trials evaluating different treatment strategies and outcomes exist and there is no general consensus on treatment although treatment to reduce hypertension and proteinuria are generally accepted. Finally, while only a few patients with this disease have received a renal allograft, to date recurrence of the disease in the allograft has not been reported.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.