
Monday
In 1927, Dr. Cecil Alport published a series on “hereditary familial congenital haemorrhagic nephritis” where he described its association with deafness and the gender differences in disease severity. https://www.ncbi.nlm.nih.gov/pubmed/20773074

Interestingly, Dr Alport initially believed the etiology of the disorder was an individual susceptibility to a toxin of an unknown organism, probably belonging to the streptococcal group. https://www.ncbi.nlm.nih.gov/pubmed/20773074

The pathogenesis of the disorder known as Alport syndrome remained unknown until early 1970’s, when advances in electron microscopy allowed the identification of characteristic abnormalities in GBMs. https://www.ncbi.nlm.nih.gov/pubmed/4343992

Dr Curtis Atkin, who suffered from Alport syndrome himself, mapped for the first time the affected gene to the long arm of the X chromosome (Xq22) in 1988. https://www.ncbi.nlm.nih.gov/pubmed/3422540

Shortly after, investigators from U of Oulu and U of Pennsylvania discovered the COL4A5 gene and mapped it to the same region known to contain the locus for X-linked AS.
https://www.ncbi.nlm.nih.gov/pubmed/1689491
https://www.ncbi.nlm.nih.gov/pubmed/2339699

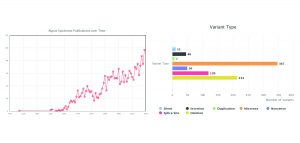
This year, close to 100 articles were published on AS and more than 800 different mutations have been identified.
https://www.ncbi.nlm.nih.gov/pubmed/20574986
https://arup.utah.edu/database/ALPORT/ALPORT_links.php
Tuesday
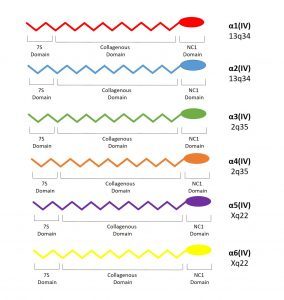
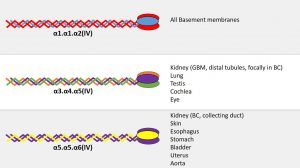
Collagen IV is only present in basement membranes and its family comprises six genetically distinct α-chains, each of which contains a collagenous, non-collagenous (NC1) and cysteine/lysine-rich amino terminal (7S) domains
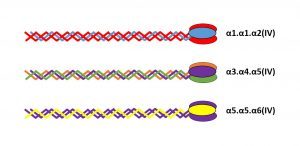
Collagen IV α-chains form heterotrimeric molecules called promoters. While 56 theoretical combinations exist, only 3 have been found in vivo: α1α1α2(IV), α3α4α5(IV) and α5α5α6(IV). https://www.ncbi.nlm.nih.gov/pubmed/18219669
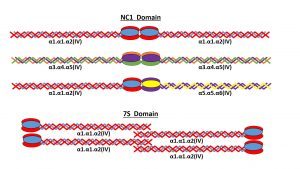
Promoters form dimers (NC1 domain) and tetrameters (7S domain) to create collagenous networks. The only 3 distinct networks found in basement membranes are: α1.α1.α2(IV)-α1.α1.α2(IV), α3.α4.α5(IV)-α3.α4.α5(IV) and α1.α1.α2(IV)-α5.α5.α6(IV)
There are significant anatomical and histological differences in the distribution of collagen IV networks, which may explain differences in clinical presentation and may be exploited for diagnostic purposes. https://www.ncbi.nlm.nih.gov/pubmed/12815141
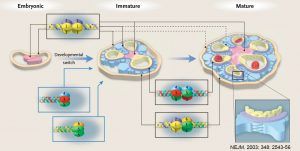
In addition to tissue-specific differences, there are temporal differences in collagen IV networks composition, as seen in early development of GBMs and BC. https://www.ncbi.nlm.nih.gov/pubmed/12815141
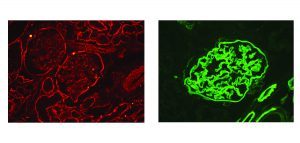
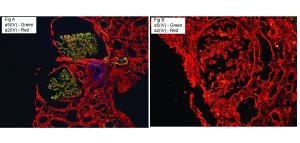
IF showing normal distribution of α2(IV) and α5(IV). α2 (Fig A) is positive in all basement membranes (mesangium is normally stronger than GBMs). α5 (Fig B) is positive in GBMs, BC, distal tubules and collecting ducts (not shown).
Wednesday
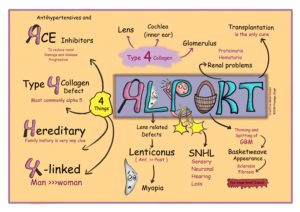
Alport syndrome (AS) is a heterogeneous disorder caused by mutations in COL4A3, COL4A4 and COL4A5 and is characterized by progressive renal disease, hearing loss and ocular defects. Image from creativemeddoses.com
AS Classification Working Group classifies the disease into 3 types: X-linked, autosomal and digenic. While controversial, it considers heterozygous mutations of COL4A3/COL4A4, and thin GBM disease as forms of AS. https://www.ncbi.nlm.nih.gov/pubmed/29551517

If AS is suspected, all 3 genes (COL4A3/4/5) should be examined. Establishing mode of inheritance is important for genetic counseling, identifying at-risk family members and potential living related donors. https://www.ncbi.nlm.nih.gov/pubmed/29987460
Women are affected more often than men by X-linked AS but are frequently under-diagnosed; yet 15-30% will develop ESRD. A high level of suspicion is critical to monitor and intervene early. https://www.ncbi.nlm.nih.gov/pubmed/27287265
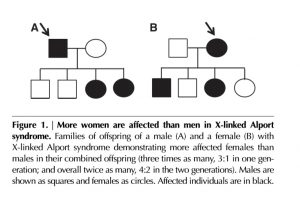
Strong genotype-phenotype correlation is seen in X-liked AS in males (Fig A), but not in females (Fig B). https://www.ncbi.nlm.nih.gov/pubmed/10752524
https://www.ncbi.nlm.nih.gov/pubmed/14514738

Other genetic and non-genetic factors may affect clinical presentation and progression. Concurrent mutations in ACTN4, NPHS2 and MYO1E are few of the known examples.
https://www.ncbi.nlm.nih.gov/pubmed/25739341
https://www.ncbi.nlm.nih.gov/pubmed/18726620
In addition to well-known manifestations of AS (hematuria, proteinuria, irregular GBMs), patients with mutations in collagen IV genes may present predominantly with NS and show FSGS lesions on biopsy. https://www.ncbi.nlm.nih.gov/pubmed/26346198

In a large cohort of patients with CKD, only 38% of patients with diagnostic variants in COL4A3/4/5 had a clinical diagnosis of AS or thin BM disease. https://www.ncbi.nlm.nih.gov/pubmed/31116938
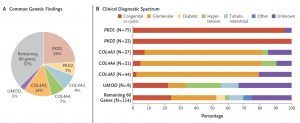
Deletions encompassing the 5′ ends of COL4A5 and COL4A6 are associated with leiomyomatosis in GI, respiratory and female GU tracts, in addition to typical renal manifestations.
https://www.ncbi.nlm.nih.gov/pubmed/21380622
https://www.ncbi.nlm.nih.gov/pubmed/29409873
Although exceedingly rare, patients with large deletions in COL4A5 may develop post-transplant anti-GBM disease.
Thursday
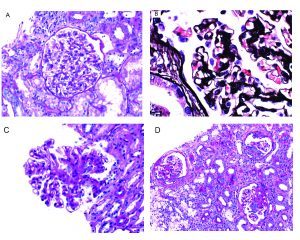
LM glomerular findings in AS are variable and include: normal glomeruli (A), irregular capillary loop contours (B), mesangial expansion with rigid loops (C), and global/segment glomerulosclerosis with epithelial cell capping (D).
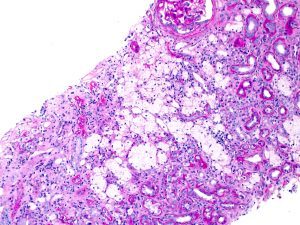
Interstitial foam cells may be seen in biopsies from patients with AS; however, this finding is not sensitive nor specific for the diagnosis and may be seen in any condition with chronic proteinuria.
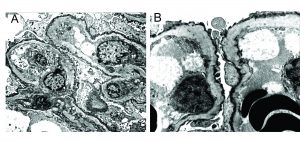
Ultrastructural evaluation of basement membranes is essential in patients with suspicion for AS. Characteristic findings include variability in GBM thickness, splitting of lamina densa, basket-weaving and irregular GBM contours.
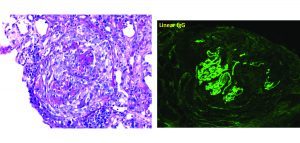
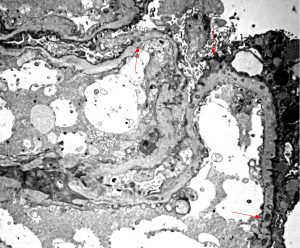
Occasionally, electron dense deposits may be seen within the mesangium or in-between the abnormal layers of GBM in patients with AS, mimicking an immune-complex mediated disease. https://www.ncbi.nlm.nih.gov/pubmed/16647952
Diffusely thin GBMs without thickening or other irregularities is within the spectrum of AS and patients should undergo genetic testing if clinical suspicion for AS exists.
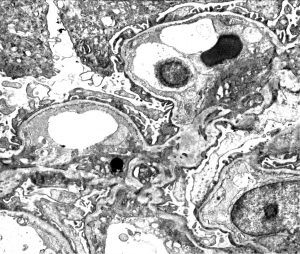
While GBM abnormalities are quite characteristic for AS, other rare genetic disorders may show overlapping features. https://www.ncbi.nlm.nih.gov/pubmed/12722046
α5(IV) IF staining may prove an abnormal distribution of collagen IV α chains in some patients; unfortunately, the test lacks sensitivity and a normal pattern does not rule out the diagnosis. Fig A: normal; Fig B: X-linked AS.
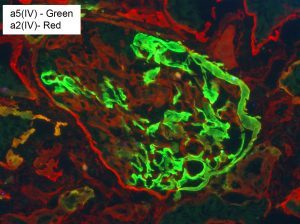
Due to random inactivation of the X-chromosome, heterozygous female patients with X-linked AS may show incomplete or discontinuous staining of capillary loops for α5(IV).
Summary
Alport syndrome (AS) is a heterogeneous genetic disorder caused by mutations in the α-chain (3, 4 or 5) of the collagen IV genes. Collagen IV is a major component of basement membranes and comprises six genetically distinct α-chains: α1(IV) through α6(IV). Human collagen IV genes are arranged in three pairs with head-to-head orientation: COL4A1 and COL4A2 (chromosome 13), COL4A3 and COL4A4 (chromosome 2) and COL4A5 and COL4A6 (X-chromosome). Each α-chain contains three domains: a short 7S N-terminal domain, a midsection collagenous domain and a C-terminal noncollagenous domain (NC1). α-chains form heterotrimeric molecules called promoters. While 56 theoretical combinations exist, only 3 have been found in vivo: α1.α1.α2(IV), α3.α4.α5(IV) and α5.α5.α6(IV). Two promoters may bind via their NC1 domain to form dimers or four may associated via their 7S domains to form tetrameters. These dimers and tetrameters create collagenous networks, which among other things, provide resistance to proteolysis. The only 3 distinct networks found in basement membranes are α1.α1.α2(IV)-α1.α1.α2(IV), α3.α4.α5(IV)-α3.α4.α5(IV) and α1.α1.α2(IV)-α5.α5.α6(IV). There are significant anatomical differences of these networks among basement membranes. The α1.α1.α2(IV)-α1.α1.α2(IV) network is ubiquitous and present in all basement membranes. The α3.α4.α5(IV)-α3.α4.α5(IV) network is present in glomerular basement membranes, basement membranes of distal tubules, focally within the Bowman’s capsule and in basement membranes of the lung, testis, cochlea, and eye. Lastly, the α1.α1.α2(IV)-α5.α5.α6(IV) network is present in the Bowman’s capsule, basement membrane of the collecting duct, epidermal basement membrane, smooth muscle cell basement membrane of the urinary bladder, uterus, esophagus, and fundus of the stomach, and vascular smooth muscle cell basement membrane of the aorta. Due to this differential composition of basement membranes, it is possible to understand the clinical presentation associated with each mutation. Due to widespread distribution of α1(IV) and α2(IV), mutations in COL4A1 and COL4A2 are incompatible with life. Mutations in COL4A3, COL4A4, and COL4A5 cause the classic AS manifestations which include progressive renal disease, hearing loss, and ocular defects. Large deletions encompassing the 5′ ends of COL4A5 and COL4A6 are associated with leiomyomatosis in the GI, respiratory and female GU tracts, in addition to typical renal manifestations. Interestingly, there are not only anatomical differences but also temporal differences in basement membrane composition. Specifically, during embryologic development of glomerular basement membranes, the α1.α1.α2(IV)-α1.α1.α2(IV) network appears at the start of capillary loop formation but is gradually replaced by the α3.α4.α5(IV)-α3.α4.α5(IV) network in the mature glomerulus.
Alport syndrome is currently classified into 3 types, according to Alport Syndrome Classification Working Group: X-linked, autosomal and digenic. It is worth noting that this new classification renders a diagnosis of AS to heterozygous female patients with COL4A5 mutations (previously considered “carriers”), as well as heterozygous male or female patients with mutations in COL4A3 or COL4A4 (autosomal dominant inheritance) if they exhibit hematuria or proteinuria, including patients who would have previously been diagnosed with thin basement membrane disease. Although the clinical presentation varies slightly based on the underlying genetic abnormality, patients with Alport syndrome usually present in childhood with hematuria, proteinuria, progressive renal insufficiency, sensorineural hearing loss, and lenticonus. Approximately 80 – 85% of patients show X-linked inheritance. While X-linked and autosomal forms with homozygous mutations have similar clinical presentation, patients may be suspected clinically to have a biallelic autosomal mutation by the presence of severe disease in young females, consanguinity in the family or presence of hematuria in the father of a male patient. On the other hand, heterozygous autosomal Alport syndrome typically has a milder clinical presentation than the X-linked type, with only a minority of patients progressing to ESRD, and those who do so are usually over 40 or 50 years of age.
While definitive diagnosis requires genetic testing, renal biopsy findings are extremely helpful, especially to rule out other glomerular diseases. By light microscopy, glomerular findings are variable and can range from normal glomeruli to global and segmental glomerulosclerosis with significant epithelial cell capping. Early findings may also include mesangial matrix expansion, rigid capillary loops, and irregular loop contours, best seen on silver-based stains. As the disease progresses, the degree of tubulointerstitial scarring also advances. Clusters of interstitial foam cells are frequently present, but this finding is not sensitive nor specific for the diagnosis and may be seen in any condition with chronic proteinuria. Routine immunofluorescence is typically negative, although non-specific trapping of immunoreactants may occasionally be observed. Ultrastructural evaluation of basement membranes is essential in patients with suspicion for AS. Characteristic findings include variability in GBM thickness, splitting of lamina densa, basket-weaving, and irregular GBM contours. Occasionally, electron-dense deposits may be seen within the mesangium or in-between the abnormal layers of GBM, mimicking an immune-complex mediated disease. Diffusely thin GBMs without thickening or other irregularities are within the spectrum of AS and patients should undergo genetic testing if clinical suspicion exists. While these GBM abnormalities are quite characteristic, other rare genetic disorders, such as those secondary to WT1 or LAMB2 mutations, may show overlapping features. Immunofluorescence staining for α-chains may provide valuable information on individual cases; however, this test lacks sensitivity and a normal pattern of staining does not rule out the diagnosis. Male patients with X-linked AS will typically have complete loss of α5(IV) stain in GBM, Bowman’s capsule, and distal tubule, while female patients may show discontinuous or incomplete staining. On the other hand, male and female patients with autosomal forms of the disease typically show complete loss of α5(IV) staining of GBM and distal tubules, with preserved staining of Bowman’s capsule. Regardless of the pattern of staining, genetic testing including all three genes: COL4A3, COL4A4, and COL4A5 should ideally be performed to establish the mode of inheritance which will be helpful in providing genetic counseling to patients, identifying at-risk family members and potential living related kidney donors.
References:
Khoshnoodi J, et al. mammalian collagen IV. Microsc Res Tech. 2008; 71: 357-370.
Hudson BG, et al. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med. 2003; 348: 2543-56.
Kashtan CE, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport Syndrome Classification Working Group. Kidney Int 2018; 93: 1045-51.
Savige J, et al. Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatr Nephrol. 2019; 34: 1175-89.
Gast C, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2016; 3: 961-70.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.