
Monday:
Day #1, Tweetorial #1:
Welcome to a #DiseaseWeek highlighting lupus nephritis from @arkanalabs. We’ll present some daily Tweetorials to dive into lupus classification, kidney pathology, and new therapies.

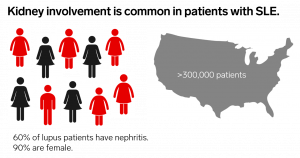
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that affects over 5 million worldwide and over 300,000 in the US alone. It has a complex etiology, involving a combination of multiple genetic and environmental factors.
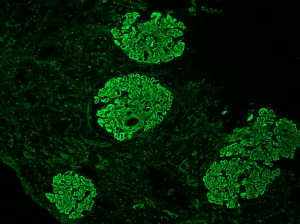
The majority of SLE patients develop nephritis. Women are disproportionately affected, as well as patients of minority populations, including African Americans, Hispanics, Asians, Native Americans, Alaska Native, Native Hawaiians, and Pacific Islanders.
The LUMINA study (Lupus in Minority Populations: Nature versus Nurture) demonstrated that Black women are more likely to have multi-system involvement, higher disease activity, and lower socioeconomic status than Whites.
https://pubmed.ncbi.nlm.nih.gov/16905579/
The disease burden is high, both personal and economic. 65% of lupus patients live with chronic pain. Between healthcare costs and lost productivity, SLE costs patients approximately $50,000 per year. This is greater for patients that progress to ESKD and require dialysis or transplantation.
https://www.lupus.org/resources/lupus-facts-and-statistics
The initial diagnostic criteria are determined by the American College of Rheumatology (ACR) and the European League against Rheumatism (EULAR). Four of 11 disease manifestations are required, which would result in quite disparate disease presentations.
https://www.ncbi.nlm.nih.gov/pubmed/9324032

Due to the heterogeneity of clinical presentations, a diagnosis of SLE is often delayed – with the average time to diagnosis being 6 years! Unfortunately, this can lead to organ damage, as patients may have benefitted from immunosuppressive therapy.
https://ard.bmj.com/content/74/Suppl_2/812.3
Renal disorder, or nephritis, is one of the 11 criteria. Therefore, the presence of nephritis was not required for a diagnosis of SLE. Additionally, nephritis was insufficient for a diagnosis of SLE, which used to require an additional 3 clinical manifestations! However, these criteria have recently changed.

Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) and the British Isles Lupus Assessment Group (BILAG) were utilized to score lupus manifestations with a point value to determine when to initiate and stop therapy. Now a similar scoring system is used for diagnosis, allowing for increased sensitivity and earlier diagnosis.
SLEDAI scoring is most commonly used in the United States. Here’s a link to the study and it can be easily assessed with this calculator by QxMD.
https://pubmed.ncbi.nlm.nih.gov/1599520/
https://qxmd.com/calculate/calculator_335/sledai-2k
The new diagnostic criteria are known as the SLICC criteria, produced by the Systemic Lupus International Collaborating Clinics. The highest score in each domain counts and a score of 10 is required for a diagnosis of SLE.
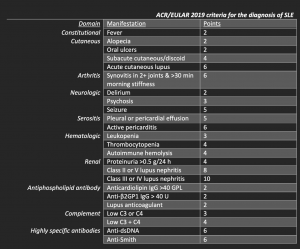
Under this new scoring system, a diagnosis of focal or diffuse lupus nephritis is sufficient for a diagnosis of SLE in patients with a positive antinuclear antibody (ANA) test. 85% of surveyed rheumatologists felt that a diagnosis of SLE could be made with a positive ANA and proliferative glomerulonephritis alone.

We’ll next examine lupus pathogenesis, so please join us for a continuation of #DiseaseWeek! /13
Day #1, Tweetorial #2:
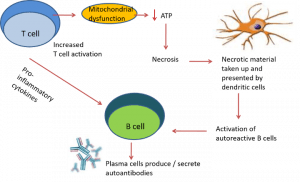
Lupus pathogenesis is caused by dysfunction in T-cell and B-cell activation. Lupus T cells show mitochondrial dysfunction and reduced generation of ATP, which predisposes to necrotic versus apoptotic cell death.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3423541/
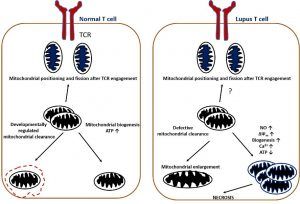
Antigen-presenting cells take up necrotic material and become activated through toll-like receptors. This leads to activation of autoreactive B cells, which differentiate into plasma cells and secrete autoantibodies (see image below).
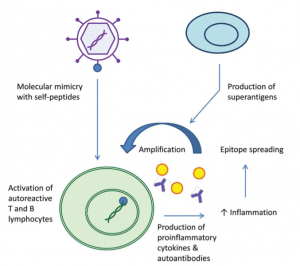
Infections may trigger autoimmunity in SLE. Molecular mimicry of microbial components with self-antigens can result in activation of autoreactive T cells. This can result in cytokine and antibody production against self-antigens.
Epitope spreading (both intramolecular and intermolecular) results in amplification of this inflammation. Additionally, superantigens (bacterial, viral) can non-specifically activate T lymphocytes, which further promotes inflammatory responses.
https://www.ncbi.nlm.nih.gov/pubmed/24471448
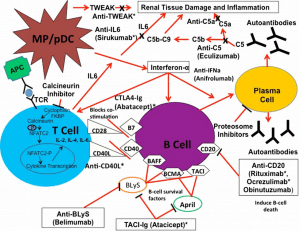
There are multiple inflammatory pathways implicated in the initiation and propagation of lupus nephritis and flares, as shown below. These can be targeted for treatment and several are under active investigation in clinical trials.
Tune in tomorrow to begin to dive into the various classes of lupus nephritis and get started with kidney pathology!
Tuesday:
Day #2, Tweetorial #1:
On day 2 of #DiseaseWeek at @arkanalabs, we’ll dive into lupus nephritis.
Let’s start with a #DiseaseWeek challenge question! What proportion of adult patients with SLE has nephritis?
- 10%
- 25%
- 50-60%
- 80%
The answer is C. 50-60% of adults with SLE will develop lupus nephritis. In children with SLE, 80% have nephritis.
https://www.ncbi.nlm.nih.gov/pubmed/26683208
https://www.ncbi.nlm.nih.gov/pubmed/23904865
In a majority of patients, renal involvement in SLE often occurs in the first year following diagnosis but can occur at any time in the disease course.
The presence of lupus nephritis increases morbidity and mortality. In a recent retrospective cohort study of 325 patients with systemic lupus erythematosus (SLE) compared to age and gender-matched population controls, SLE patients with nephritis have more than two-fold higher mortality than SLE patients without nephritis.
https://www.ncbi.nlm.nih.gov/pubmed/31072277
Double-stranded DNA antibody titers help predict lupus nephritis flares if they are present. Double-stranded DNA antibodies are often seen in patients with lupus nephritis and correlate with disease activity.
An increase in dsDNA antibody titer is associated with an increased risk of renal flare and with hypocomplementemia (with an inverse correlation with C3). Antibody titers drop in response to treatment, with a 50% decrease in dsDNA antibody titer reducing renal flare rate 52-53% in two treatment cohorts.
https://www.ncbi.nlm.nih.gov/pubmed/15818711
Anti-Smith antibodies are also highly specific for SLE, but have poor sensitivity (29%). Serum anti-Smith antibodies correlate with membranous lupus nephritis.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1003610/

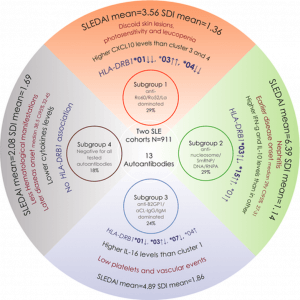
Four subgroups of SLE have recently been described according to autoantibody profiles, HLA haplotypes (at HLA-DRB1 locus), cytokine profiles, and clinical associations. Patients with lupus nephritis, on average, have higher disease activity, earlier disease onset, and an autoantibody profile with anti-nucleosome/anti-SmRNP/anti-DNA, and anti-RNPA antibodies.
https://pubmed.ncbi.nlm.nih.gov/34658170/
Day #2, Tweetorial #2:
Time for another #DiseaseWeek tweetorial. How do we classify lupus nephritis?
The International Society of Nephrology (ISN) and Renal Pathology Society (RPS) created a lupus nephritis classification system consisting of 6 classes of disease based on morphologic findings on kidney biopsy. A simplified view is below.

The classes are based on the distribution of immune deposits and the proportions of glomeruli affected. This classification is VERY glomerulocentric.

Here’s an overview of the classification of lupus nephritis.
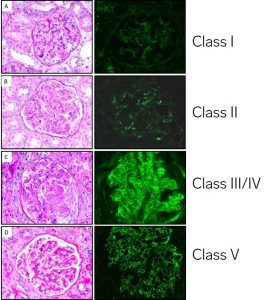
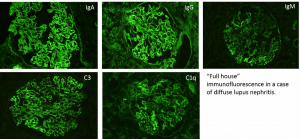
In class I (minimal mesangial) lupus nephritis, mesangial immune complexes are identified by immunofluorescence without any significant changes by light microscopy. IgG deposits are necessary, but there is sometimes not ‘full house’ staining. #DiseaseWeek /4
‘Full house’ staining is when there are all 3 immunoglobulin heavy chains (IgA, IgG, and IgM) and both complement components routinely stained for (C3 and C1q), along with light chains lighting up on kidney biopsy. #DiseaseWeek/5
In class II (mesangial proliferative) lupus nephritis, mesangial immune complexes are present by immunofluorescence with mesangial expansion and mesangial hypercellularity by light microscopy.
Class III and IV lupus nephritis show proliferative changes within glomeruli. In class III lupus nephritis (focal lupus nephritis), less than 50% of glomeruli show proliferative lesions. In class IV lupus nephritis, greater than 50% of glomeruli contain proliferative changes (diffuse lupus nephritis).
Focal (class III) or diffuse (class IV) lupus nephritis can have concurrent subepithelial deposits. If >50% of the glomerular capillary loops are involved, there is concurrent membranous lupus nephritis (class V lupus nephritis). We’ll dive more into membranous lupus nephritis tomorrow.
If there are subepithelial IgG deposits without proliferative lesions, there is a ‘pure’ membranous lupus nephritis (class V).
When there is glomerular immune deposition with >90% of glomeruli being globally sclerotic, there is advanced sclerosing lupus nephritis (class VI), which is end-stage kidney disease due to lupus.
Let’s do another #DiseaseWeek challenge question! Which of the following lupus classifications can be seen in the same biopsy?
- ISN/RPS class II + V
- ISN/RPS class III + IV
- ISN/RPS class V + VI
- ISN/RPS class III + V
The answer is (D), ISN/RPS class III + V. Concurrent membranous lupus nephritis (ISN/RPS class V) can be diagnosed with proliferative lupus nephritis (class III or IV) if >50% of the glomerular capillary loops in >50% of glomeruli show subepithelial and/or intramembranous IgG deposits.
Tune in tomorrow as we continue #DiseaseWeek with a discussion of the NIH activity and chronicity indices to evaluate lupus nephritis!
Wednesday:
Day #3, Tweetorial #1:
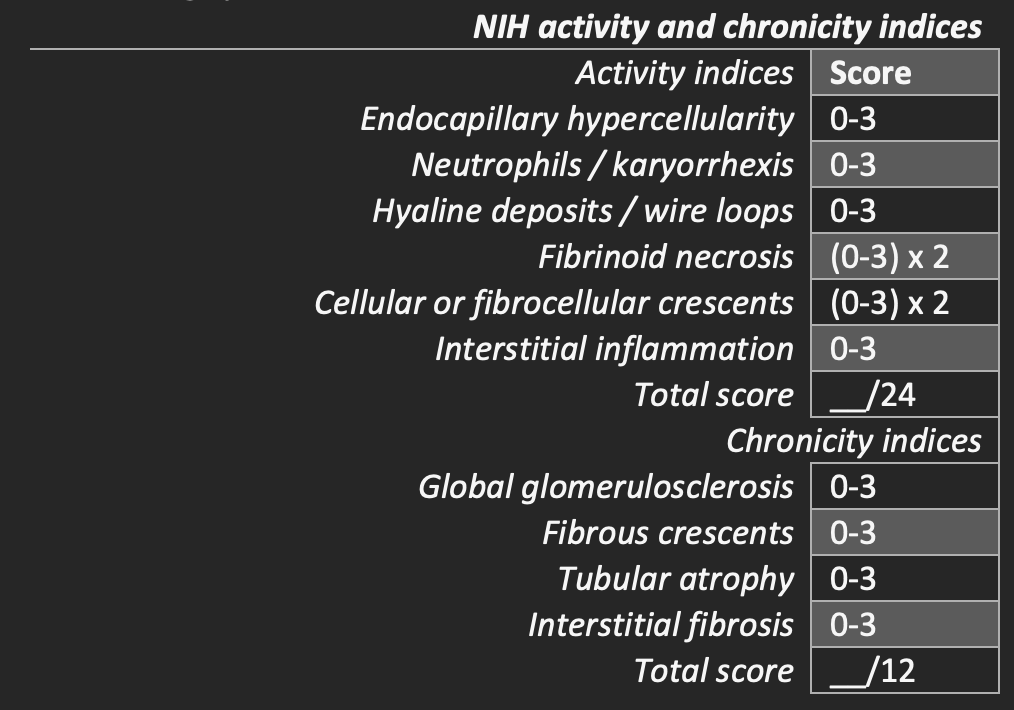
A semi-quantitative grading system of pathologic features on kidney biopsies allows for monitoring response to treatment and showing disease progression. This grading system to assess activity and chronicity is the modified NIH lupus nephritis activity and chronicity indices.

Indicators of disease activity include endocapillary hypercellularity, neutrophils or karyorrhexis within glomerular capillary loops, fibrinoid necrosis, hyaline deposits, cellular or fibrocellular crescents, and interstitial inflammation.
Crescents and fibrinoid necrosis are weighted twice as they have a worse impact on prognosis.
The scoring is based on the percentage of glomeruli with each feature in the biopsy on a 0 to 3 scale, with a score of 0 = not present, 1 = <25% glomeruli, 2 = 25-50% glomeruli, and 3 indicating >50% glomeruli.
Indicators of disease chronicity include the total percentage of global glomerulosclerosis, fibrous crescents, tubular atrophy, and interstitial fibrosis.
Grading the NIH activity and chronicity indices allow comparison in repeat biopsies, as well as for standardized assessment for lupus clinical trials. Reporting of these indices is recommended on all kidney biopsies for patients with focal or diffuse lupus nephritis.
https://www.ncbi.nlm.nih.gov/pubmed/29459092
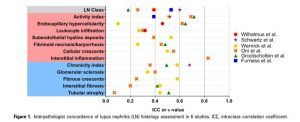
While important to assess activity and chronicity, there is high interobserver variability. In a review of 6 studies of 4+ pathologists studying interobserver agreement, the kappa statistics were 0.52 and 0.49 and only 0.325 for the ISN/RPS disease class. Improvement of this classification is still needed.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6829183/
Day #3, Tweetorial #2:
For today’s continuation of lupus nephritis #DiseaseWeek at @arkanalabs, we’ll discuss membranous lupus nephritis.
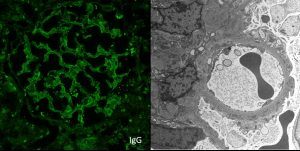
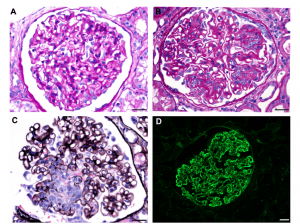
The image shows granular mesangial and capillary loop staining for IgG with corresponding subepithelial, intramembranous, and mesangial electron-dense deposits identified on electron microscopy. These are typical features of membranous lupus nephritis.
Patients with membranous lupus nephritis present with proteinuria. Unlike primary membranous glomerulopathy, there is an increased frequency of subnephrotic proteinuria, with 40% of patients having < 3 g proteinuria/day and 20% with less than 1 g/day.
Patients with membranous lupus nephritis are less likely to show active serologies (anti-dsDNA, anti-Smith antibodies, high titer ANA), hypertension, and renal insufficiency when compared to patients with focal or diffuse lupus nephritis.
One-third of patients with membranous lupus nephritis have isolated kidney disease prior to other systemic manifestations, which may develop later.
On a kidney biopsy, membranous lupus nephritis can look like a ‘primary’ membranous glomerulopathy. By light microscopy, the glomerular capillary loops are thickened, with ‘spikes’ or ‘holes’ seen along with the glomerular capillary loops on silver stains.
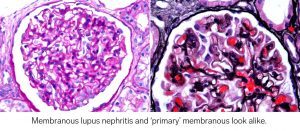
If a patient does not have a pre-existing diagnosis of SLE, what would push you towards a diagnosis of membranous lupus nephritis versus a primary membranous glomerulopathy? There was a nice study in CJASN by Kudose et al that explored this.

“Tissue ANA” is another feature, although has poor sensitivity. Tubular epithelial cell (and occasionally podocyte nuclei) show staining for IgG by routine immunofluorescence, as shown below.
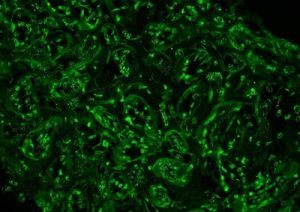
In ‘primary’ membranous glomerulopathy, IgG deposits within glomeruli are frequently IgG4-restricted. This pattern is not typically seen in MLN, where multiple IgG subclasses are often expressed (often IgG1, IgG2, and IgG3).
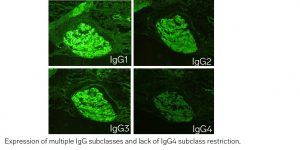
Vascular staining in membranous glomerulopathy could also indicate membranous lupus nephritis. Check out Dr. Bourne’s teaching point at https://www.arkanalabs.com/teaching-points-6/
In summary, these features that would push you towards membranous lupus nephritis are:
- Mesangial deposits
- Subendothelial deposits
- C1q deposits
- “Full house” immunofluorescence
- TBM deposits
- Tissue ANA
- Tubuloreticular inclusions
- Multiple IgG subclasses
- Vascular deposits
In SLE patients without concurrent proliferative lupus nephritis, the differential diagnosis of membranous lupus nephritis includes other secondary causes of membranous glomerulopathy, including drug rxns, infections (including Hepatitis B + C), and malignancy.
A majority of the autoantigens driving diseases in membranous lupus nephritis are unknown (approximately 2/3). To date, there are 3 autoantigens enriched in membranous lupus nephritis over primary membranous glomerulopathy. Unlike ‘primary’ membranous, PLA2R positivity is rare in membranous lupus nephritis.
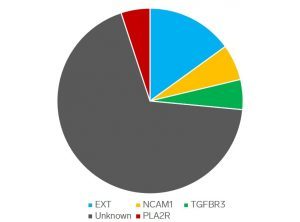
Laser capture microdissection studies have revealed the exostosin 1/2 complex (EXT) as an autoantigen in approximately 1/3 of cases of membranous lupus nephritis, and in fewer cases of primary idiopathic membranous glomerulopathy.
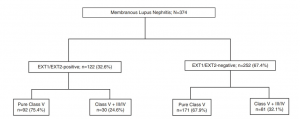
Of all membranous lupus nephritis cases, 32.6% are EXT1/2-positive. Approximately one-quarter of patients had a concurrent proliferative component (class III or IV disease).
Patients with EXT1/2-positive membranous lupus nephritis have better outcomes than EXT1/2-negative patients, including those with concurrent class III or IV lupus nephritis.
https://pubmed.ncbi.nlm.nih.gov/33478971/
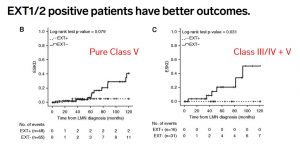
This work has been replicated in independent cohorts, and EXT1/2-positive membranous lupus nephritis shows reduced creatinine, higher GFR, and increased rates of remission compared to EXT1/2-negative membranous lupus nephritis patients.
https://pubmed.ncbi.nlm.nih.gov/34307993/
Neural cell adhesion molecule 1 (NCAM1) and Transforming growth factor-beta receptor 3 (TGFBR3) are two additional targets in membranous lupus nephritis. Protein G immunoprecipitation was used to elute immune complexes from biopsy tissue for target antigen discovery.
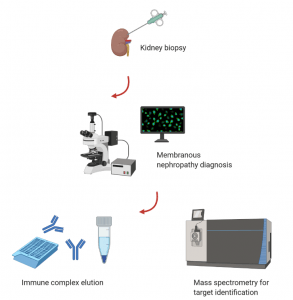
NCAM1 was recently identified as a target antigen in membranous lupus nephritis with a frequency of 6.6% of total cases. Patients with NCAM1-positive membranous nephropathy were predominantly young women, presented with proteinuria, and had a history of SLE.
https://pubmed.ncbi.nlm.nih.gov/33045259/
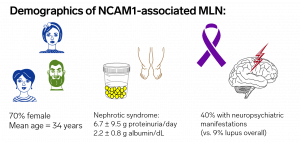
Patients with NCAM1-associated membranous lupus nephritis can have a concurrent proliferative component with concurrent focal or diffuse lupus nephritis (class III or IV + V).
https://pubmed.ncbi.nlm.nih.gov/33045259/
Transforming growth factor-beta receptor 3 (TGFBR3) positivity is present in 5.5% of cases of membranous lupus nephritis. Similar to EXT1/2-positive membranous and NCAM1-positive membranous, it can be seen with our without a proliferative component (class III or IV disease).
Patients with EXT1/2, NCAM1, or TGFBR3-positive membranous are of a younger age than patients with ‘primary’ membranous nephropathy. Positivity of any of these 3 antigens is much more common in the setting of membranous lupus nephritis than primary membranous nephropathy.

Thanks for tuning in with us for some discussion on membranous lupus nephritis at #DiseaseWeek.

Thursday:
Day #4, Tweetorial #1:
While glomerular lesions are the focus of lupus classification and activity scoring, vascular involvement, tubulointerstitial involvement, and concominant autoimmune disease greatly impacts prognosis.
The spectrum of renal vascular lesions in lupus nephritis includes uncomplicated vascular immune deposits, lupus vasculopathy, lupus vasculitis, thrombotic microangiopathy (including HUS/TTP), malignant hypertensive vascular lesions, and arteriosclerosis or arteriolosclerosis. #DiseaseWeek /2
https://www.ncbi.nlm.nih.gov/pubmed/9352738
https://pubmed.ncbi.nlm.nih.gov/26022040/
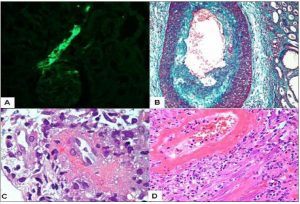
The presence of renal vascular lesions predicts a poor prognosis. The 10-year survival is reduced from 85.9% (lupus nephritis without vascular lesions) to 58.0% (lupus nephritis with vascular lesions).
https://www.ncbi.nlm.nih.gov/pubmed/1867181
In these electron photomicrographs of a patient with lupus nephritis and chronic thrombotic microangiopathy, subendothelial widening and glomerular basement membrane duplication are shown.
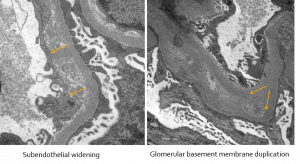
In patients with membranous lupus nephritis, similar to idiopathic membranous glomerulopathy, there is an increased risk of renal vein thrombosis and pulmonary emboli due to the hypercoagulable state of nephrotic syndrome.
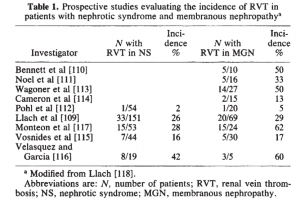
The risk of renal vein thrombosis in membranous glomerulopathy has been reported to be 5-62% in separate case series, as shown below.
https://www.ncbi.nlm.nih.gov/pubmed/3906225
The etiology of renal vein thrombosis in nephrotic syndrome is due to urinary losses of circulating anticoagulants, including antithrombin 3. There is increased plasma fibrinogen, clotting factors (factor V, VII, VIII, and X), and thromboplastin that promote thrombosis. Thrombocytosis is also common.
https://www.ncbi.nlm.nih.gov/pubmed/5578557
Signs of a renal vein thrombosis on a kidney biopsy include:
- RBC congestion or neutrophil margination in glomerular capillaries
- Focal glomerular capillary loop fibrin thrombi
- Diffuse interstitial edema
- Disproportionate interstitial fibrosis and tubular atrophy to the degree of global glomerulosclerosis
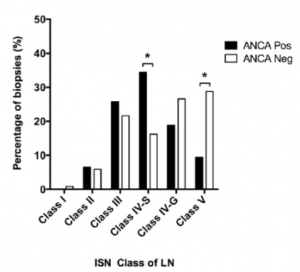
Another etiology of vascular disease in the setting of lupus nephritis is concurrent ANCA-associated glomerulonephritis. 12.6% of patients with SLE have positive serologies for antineutrophil cytoplasmic antibodies (ANCA). ANCA seropositivity is associated with a segmental distribution within glomeruli in diffuse lupus nephritis.
https://www.ncbi.nlm.nih.gov/pubmed/28750930
Lupus nephritis patients with a positive ANCA serology most commonly have anti-MPO antibodies (82%). Of the remaining patients, 11% had dual MPO and PR3 antibodies, and 7% had PR3 antibodies.
https://www.ncbi.nlm.nih.gov/pubmed/28750930
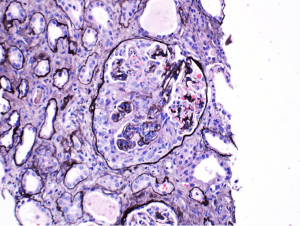
Lupus nephritis with a positive ANCA serology are more likely to have active necrotizing lesions.
https://www.ncbi.nlm.nih.gov/pubmed/28750930
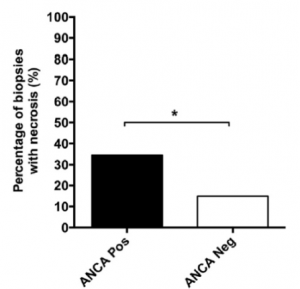
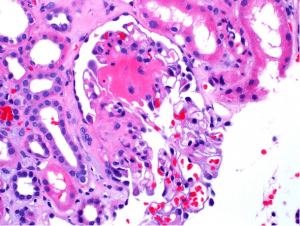
ANCA positivity in diffuse lupus nephritis is more likely to have a pauci-immune phenotype with fewer immune deposits than without ANCA positivity. Lupus patients with ANCA and class IV-S nephritis show a worse baseline renal function and have more active lupus serologies (including increased anti-dsDNA titers).
https://www.ncbi.nlm.nih.gov/pubmed/29055427
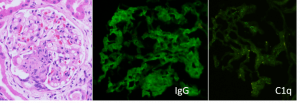
An additional concurrent vascular disease that can occur in lupus patients is due to anti-phospholipid antibody syndrome, which causes arterial (but not venous) thrombosis. Up to 30% of kidney biopsies of SLE patients have antiphospholipid antibody-induced (APL) nephropathy, with or without lupus nephritis.
https://www.ncbi.nlm.nih.gov/pubmed/11752020
Antiphospholipid antibody (APL) nephropathy can present clinically with hypertension (mild, accelerated, or malignant), chronic renal failure, hematuria, +/- proteinuria. APL nephropathy is associated with lupus anticoagulants, but not anticardiolipin antibodies.
https://pubmed.ncbi.nlm.nih.gov/26022040/
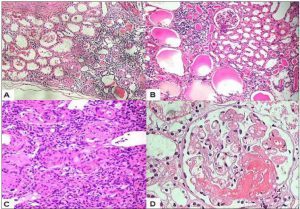
APL nephropathy presents as a spectrum of lesions within kidney biopsies, including thrombotic microangiopathy (with glomerular and vascular involvement), fibrous or mucoid intimal hyperplasia, organizing thromboses with fibrous or fibrocellular luminal stenosis, and disproportionate interstitial fibrosis and tubular atrophy due to ischemic injury.
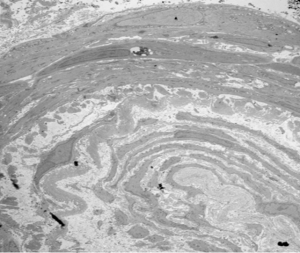
Here’s one example (of many possible disease manifestations) – Onion-skinning-like reaction due to concentric myointimal proliferation within an arteriole, seen on electron microscopy.
Day #4, Tweetorial #2:
Acute tubulointerstitial nephritis can be a disease manifestation of SLE and is a component of the NIH activity index, although only scored up to 3 of the 24 activity points. Up to 25% interstitial inflammation is scored as (1), 25-50% as (2), and >50% as (3).
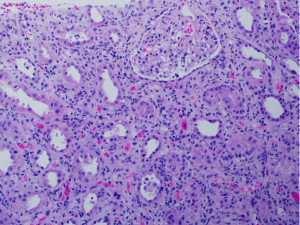
Concurrent tubulointerstitial disease can occur in any type of lupus nephritis but is most common in class III and IV disease. Tubulointerstitial disease includes interstitial inflammation, edema, and mixed infiltrates. Chronic tubulointerstitial disease results in interstitial fibrosis and tubular atrophy.
Immune deposits can occur along tubular basement membranes. There is no correlation between the degree of deposition and inflammation/scarring.
https://www.ncbi.nlm.nih.gov/pubmed/3540691
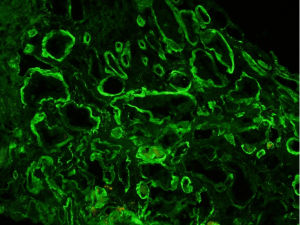
Tubulointerstitial lesions (interstitial inflammation of which includes tubulitis) in SLE is an important predictor of kidney outcomes in patients with lupus nephritis.
Day #4, Tweetorial #3:
Another kidney disease in SLE patients that is not part of the standard classification, but contributes to renal dysfunction and proteinuria is lupus podocytopathy. Lupus podocytopathy is seen in 1-2% of biopsies in patients with systemic lupus erythematosus.

Lupus podocytopathy is diagnosed clinically by meeting clinical criteria for lupus (established by the American College of Rheumatology), the presence of nephrotic syndrome, and severe to global podocyte foot process effacement.
Lupus podocytopathy can be seen with negative immunofluorescence, or with mesangial immune deposition, which can be accompanied by mesangial proliferation on light microscopy.
https://www.ncbi.nlm.nih.gov/pubmed/26983708
Therefore, it can be seen without a diagnosis of lupus nephritis, with minimal mesangial lupus nephritis (class I lupus nephritis), or with mesangial proliferative lupus nephritis (class II lupus nephritis).
The criteria for lupus podocytopathy are shown below:
https://www.ncbi.nlm.nih.gov/pubmed/26983708

A diagnosis of minimal change disease in a lupus patient is lupus podocytopathy. A majority of patients (94%) with lupus podocytopathy respond to immunosuppressive therapy with corticosteroids, however, 59.6% relapse at 5 years.
https://www.ncbi.nlm.nih.gov/pubmed/26983707
Collapsing glomerulopathy (CG) may be an extreme form of lupus podocytopathy, with worse outcomes. CG in SLE patients is associated with APOL1 risk alleles in African American patients.
https://www.ncbi.nlm.nih.gov/pubmed/23520206
Time for a #DiseaseWeek challenge question! Which of the following cases is not a lupus podocytopathy?
- ISN/RPS class II + 70% foot process effacement
- Collapsing glomerulopathy in a patient with SLE
- No immune deposits with 100% foot process effacement
- ISN/RPS class V with 70% foot process effacement
The answer is D. ISN/RPS class V is membranous lupus nephritis. A diagnosis of lupus podocytopathy excludes proliferative lesions (ISN/RPS class III or IV) and membranous glomerulopathy.
Join us tomorrow as we wrap up lupus nephritis #DiseaseWeek at @arkanalabs !
Friday:
Day #5, Tweetorial #1:
When to pursue a kidney biopsy in lupus patients is somewhat controversial. The most common indications include:
- Proteinuria >500 mg/24 h or >0.5 g/g on a urine protein-to-creatinine ratio
- Hematuria
- Active urinary sediment with cellular casts.
Determining when to stop immunosuppression for proliferative lupus nephritis can be a difficult challenge for nephrologists and rheumatologists. A prospective observational study examined kidney biopsies following maintenance immunosuppression to address this.
https://www.ncbi.nlm.nih.gov/pubmed/30045812
The study utilized the NIH activity and chronicity indices to evaluate for residual disease on biopsies. In patients with a residual NIH activity greater than 2, there was a risk of disease flare following cessation of immunosuppression.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6291576/
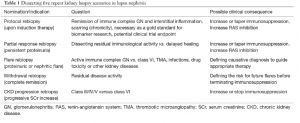
Day #5, Tweetorial #2:
Let’s explore the treatment of lupus nephritis. The nephrologists tuning in are the experts here, so please share your experiences!
Cytotoxic agents, including intravenous cyclophosphamide, has been shown to be superior to high-dose steroids for the treatment of proliferative lupus nephritis and decrease the risk of end-stage kidney disease.
https://www.ncbi.nlm.nih.gov/pubmed/3511372
IV methylprednisolone + IV cyclophosphamide increases treatment efficacy. 29% of lupus nephritis patients achieved remission with IV methylprednisolone, and 62% with cyclophosphamide, but 85% remitted using combination therapy.
https://www.ncbi.nlm.nih.gov/pubmed/8815753
There were superior outcomes with induction, followed by maintenance therapy. Induction therapy includes either cyclophosphamide or mycophenolate mofetil, while maintenance therapy is with azathioprine and prednisone.
https://www.ncbi.nlm.nih.gov/pubmed/8815753
Mycophenolate mofetil (MMF) and methylprednisolone were found to be as effective as IV cyclophosphamide and prednisolone. African American and Hispanic patients had better outcomes with MMF compared to Caucasian patients.
https://www.ncbi.nlm.nih.gov/pubmed/19369404
The first biological therapy for lupus nephritis is Belimumab, a monoclonal ab that inhibits the B-cell stimulator BAFF. When added to standard therapy, belimumab reduced disease activity, reduced severe flares, and was well tolerated.
https://www.ncbi.nlm.nih.gov/pubmed/21296403
A nice timeline of trials in treatment of lupus nephritis can be found on @RenalFellowNtwk by @DiMiRenalMD at: https://www.renalfellow.org/2019/01/09/landmark-trials-in-lupus-nephritis-look-how-far-weve-come/

There are some recent positive trials in lupus nephritis! These were nicely reviewed at #KidneyWk this year by Dr. Lightstone (shown below) and included B-cell depletion with Obinutuzumab (NOBILITY study), BAFF-APRIL blockade with Belimumab (BLISS-LN study), and calcineurin inhibitor Voclosporin (AURORA study).

The results of the BLISS-LN and AURORA studies are published and the links are below. #DiseaseWeek/9
Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis – PubMed (nih.gov)
There are several additional targets in the pipeline and hopefully more to come . . . #DiseaseWeek
Day #5 – Tweetorial #3
Recurrence of lupus nephritis is rare in allograft kidneys of SLE patients undergoing transplantation. This is largely because the same immunosuppressive drugs for transplant tolerance are used to manage lupus disease activity and flares.
For lupus nephritis patients who develop end-stage renal disease and undergo transplantation, 2.44% have disease recurrence in the allograft kidney.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3152228/
Patients at increased risk of disease recurrence include females, age < 33 years, and non-Hispanic black race. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3152228/
Although recurrent lupus nephritis is rare, it is typically aggressive, with graft failure in 93% of patients with recurrent disease.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3152228/
Summary: Lupus Nephritis
New SLE diagnostic criteria increase sensitivity for lupus nephritis diagnosis, and novel biomarkers could prove helpful to compensate for reduced specificity.
Under the new criteria established by the Systemic Lupus International Collaborating Clinic as a joint effort from the American College of Rheumatology and European League Against Rheumatism, a diagnosis of lupus nephritis can now be made with a positive antinuclear antibody and class III or IV lupus nephritis alone. This increased the sensitivity of diagnosis but reduced specificity when compared to the 1997 revision of the 1982 American College of Rheumatology SLE diagnostic criteria (Rijnink et al, 2018). With the current classification, lupus-like conditions, such as HIV-associated immune complex disease of the kidney (HIVICK) could be included in a diagnosis of SLE. The development of biomarkers is needed to differentiate lupus nephritis from other lupus-like conditions, such as infections.
The ISN/RPS classification is used to evaluate lupus nephritis on a kidney biopsy. The NIH activity and chronicity indices provide comparisons of disease progression and response to therapy.
The International Society of Nephrology/ Renal Pathology Society classification separates categories of lupus nephritis but does not provide prognostic value. The NIH Disease Activity and Chronicity indices is used in parallel to do this, but at present, there are no clear cutoffs of activity or chronicity that indicate when to treat lupus nephritis patients (Bajema et al, 2018). Establishing disease activity (and/or chronicity) cutoffs to guide when to start immunosuppressive therapy will be important. There are also likely to be subcategories of proliferative lupus nephritis that may have clinical and prognostic significance. One example are the segmental necrotizing lesions in patients with focal or diffuse lupus nephritis in patients with concurrent positive ANCA serology that predict poorer renal outcomes. The classification of lupus nephritis is also limited by glomerulocentricity, since severe tubulointerstitial injury and vascular involvement have a poor renal prognosis but are not included in the classification scheme.
A lupus classification based on the antigenic drivers of disease may exist in the future.
It is possible that lupus nephritis may be classified according to the inciting autoantigen in the future. Laser capture microdissection studies and elution of immune complexes from tissue combined with proteonomic techniques can be used to identify autoantigens within immune complex-mediated diseases. Recent examples include the exostosin 1/2 complex, which was recently identified in membranous lupus nephritis (Sethi et al, 2019), as well as neural epidermal growth factor-like 1 (NCAM1, Caza et al, 2021), and transforming growth factor-beta receptor 3 (TGFBR3, Caza et al, 2021). There are likely several other autoantigenic drivers that have yet to be identified and could represent distinct forms of the disease. Development of serological assays could allow for monitoring of disease progression and response to therapy, as is available with the two current antigens for idiopathic membranous glomerulopathy (PLA2R and THSD7A).
Improved serum and urine biomarkers could have predictive value and inform treatment.
Non-invasive serum and urine biomarkers are needed to predict the onset of lupus nephritis in patients with SLE and to predict flares for patients with established disease. Currently, SLE patients are monitored by nephrologists and rheumatologists by non-specific laboratory tests, including serum creatinine, estimated glomerular filtration rate, and urine studies (urine sediment analysis, proteinuria, and hematuria), serum complements, and serologic studies. Anti-dsDNA titers, if present, can be helpful to predict flare, as well as low serum complements. Proteinuria at one year is the best predictor, at present, for long-term kidney outcomes. However, resolution of serum creatinine and reduction of proteinuria does not always correlate with the absence of disease activity. Kidney biopsy studies have shown that residual histologic activity is present despite improved serum creatinine and/or proteinuria. Due to this, repeat kidney biopsies may be useful post-treatment to determine when to stop immunosuppressive therapy. An NIH disease activity score >2 on a post-treatment biopsy indicates poor outcomes.
There is a need for development and validation for serum and urine biomarkers to predict disease flare and response to therapy. Prospective studies evaluating novel analytes in clinical trials with long-time follow-up will be helpful to stratify risk, predict flare, monitor response to therapy, and predict disease prognosis. Molecular analyses of kidney biopsies could also be helpful in this process (Parikh et al, 2015)
Enrollment in clinical trials may establish new treatment targets and provide hope to patients at risk of renal failure.
Some factors associated with poor outcomes in lupus nephritis include African or Hispanic ethnicity, male sex, earlier age of onset, frequent disease relapses, incomplete remissions post-immunosuppressive therapy, proteinuria >4 grams at diagnosis, presence of anti-phospholipid antibodies, high titer dsDNA antibodies, anti-C1q antibodies, and crescentic disease or vascular involvement on a kidney biopsy (Rovin et al, 2019). Optimal management for many of these at-risk patients has yet to be determined. One example of improved treatment in an at-risk group is mycophenolate mofetil for proliferative lupus nephritis, which has shown improved outcomes in African American patients. Future studies are needed to determine how to optimally treat patients in these risk groups. Increased identification and enrollment in clinical trials will help evaluate the efficacy of new targets and to optimize therapy.
References:
Bajema IM, Wihelmus S, Alpers CE, et al. Revision of the International Society Classification for Lupus Nephritis: Clarification of Definitions, and Modified National Institutes of Health Activity and Chronicity Indices. Kidney International 2018; 93: 789-796.
Parikh SV, Malvar A, Song H, et al. Characterizing the Immune Profile of the Kidney Biopsy at Lupus Nephritis Flare Differentiates Early Treatment Responders from Non-Responders. Lupus Sci Med 2015; 2: e000112.
Rijnink EC, Teng YKO, Kraaji T, et al. Validation of the Systemic Lupus International Collaborating Clinics Classification Criteria in a Cohort of Patients with Full House Glomerular Deposits. Kidney International 2018; 93: 214-220.
Rovin BH, Caster DJ, Cattran DC, Gibson KL, Hogan JJ, Moeller MJ, Roccatello D, Cheung M, Wheeler DC, Winkelmayer WC, Floege J. Management and Treatment of Glomerular Diseases (Part 2): Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney International 2019; 95: 281-295.
Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravidran A, Hummel AM, Specks U, Fervenza FC, Ronco P. Exostosin 1/exostosin 2-associated membranous nephropathy. Journal of American Society of Nephrology 2019 Jun (6): 1123-1136.
Caza TN, Hassen SI, Kuperman M, Sharma SG, Dvanajscak Z, Arthur J, Edmondson R, Storey A, Herzog C, Kenan DJ, Larsen CP. Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis. Kidney International 2021 July; 100 (1): 171-181.
Caza TN, Hassen SI, Kenan DJ, Storey A, Arthur JM, Herzog C, Edmondson RD, Bourne TD, Beck Jr LH, Larsen CP. Transforming growth factor beta receptor 3 (TGFBR3)-associated membranous nephropathy. Kidney 360 2021 Aug; 2 (8): 1275-1286.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.