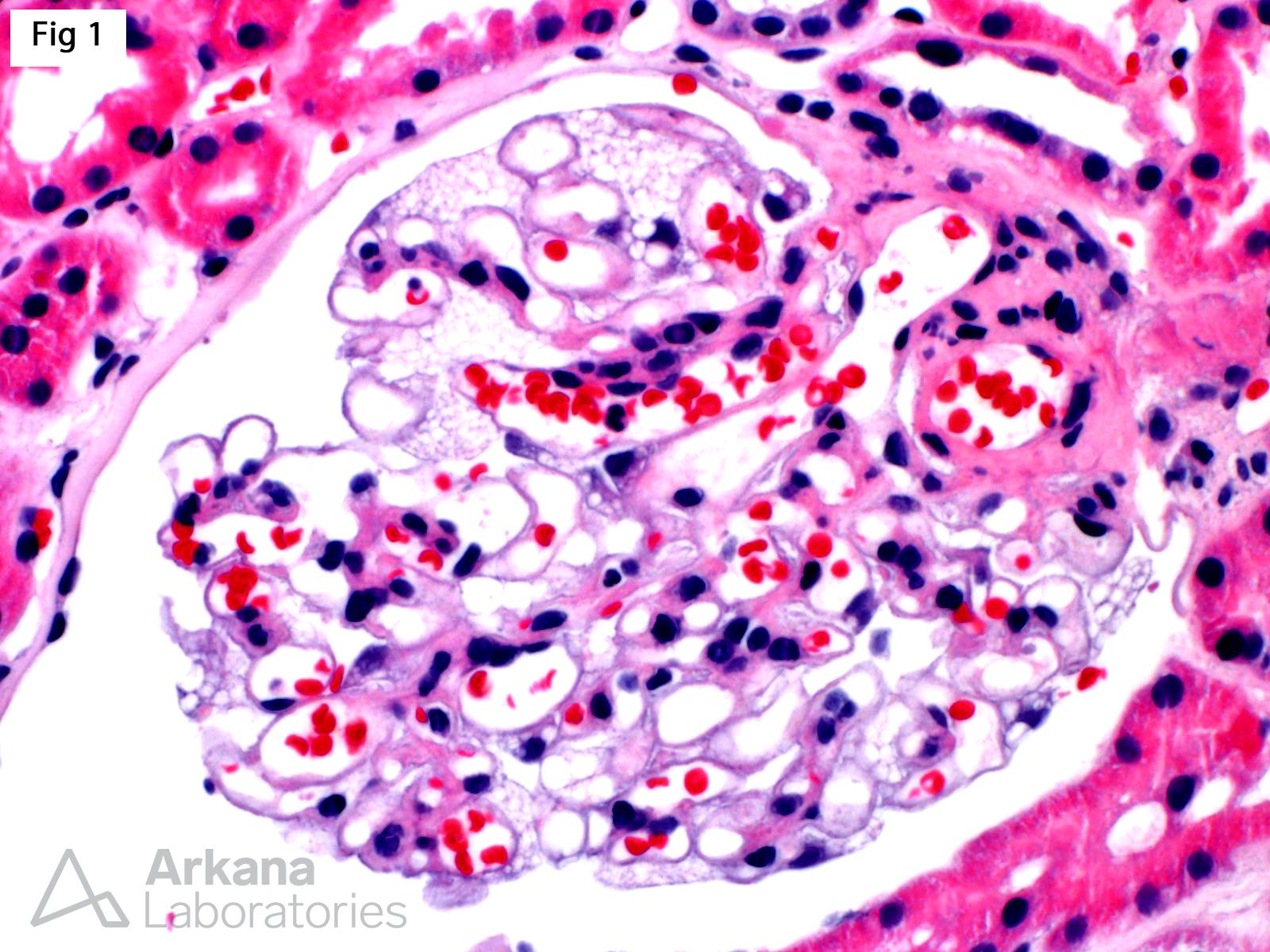
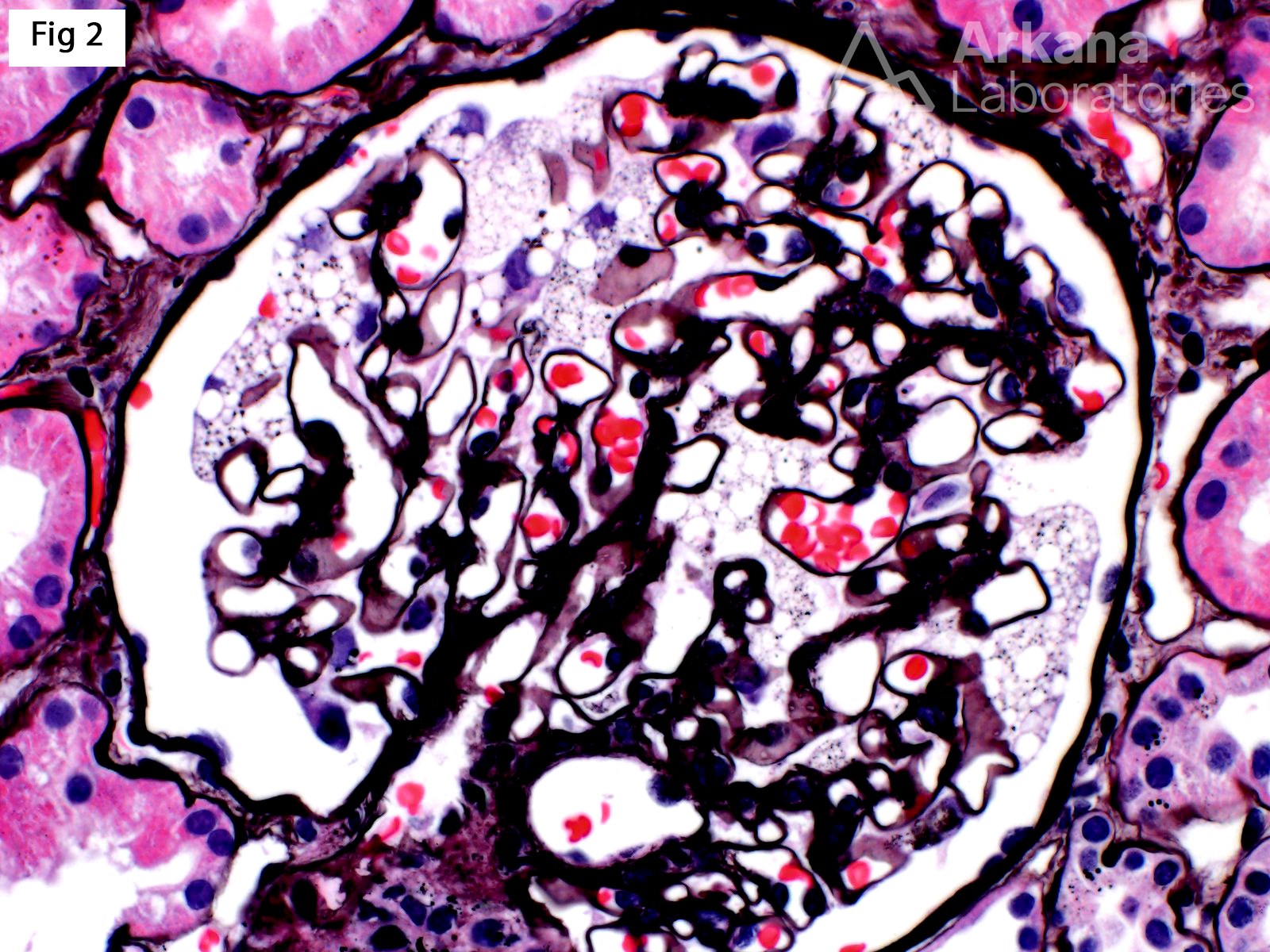
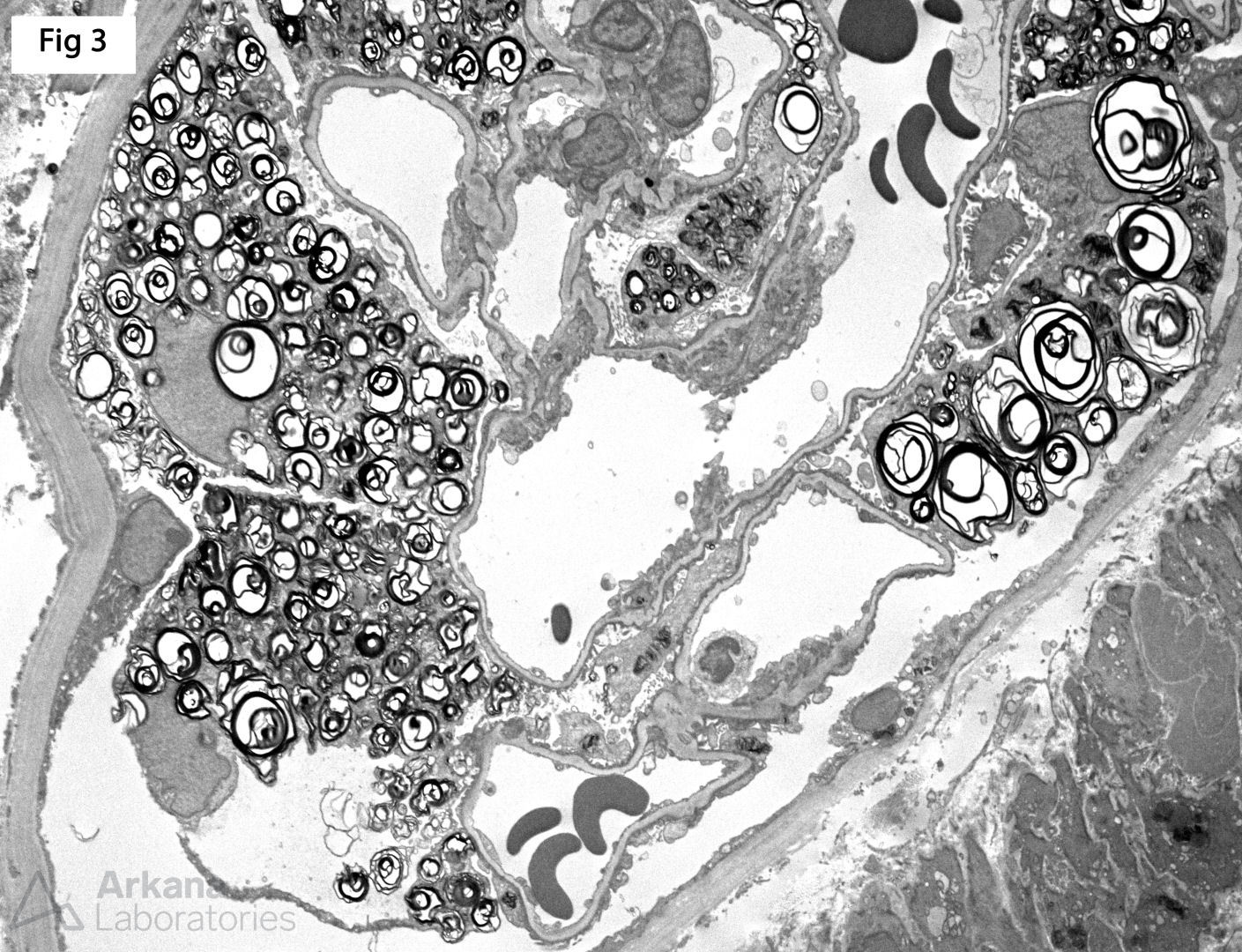
A renal biopsy was performed on a 51-year-old female with strong family history of Fabry disease and a known carrier of an alpha-glycosidase gene (GLA) pathogenic variant. At the time of the biopsy, the patient had no renal or extrarenal manifestations. The biopsy shows glomeruli with prominent visceral epithelial cells (podocytes) displaying ample vacuolated cytoplasm (Fig 1, H&E; Fig 2, Jones). Furthermore, electron microscopy shows numerous large, lamellated lipid vacuoles (myeloid bodies or zebra bodies) within the podocytes, characteristic of Fabry disease (Fig 3). Fabry disease is an X-linked disorder caused by GLA mutation and the resulting deficiency of the lysosomal enzyme α-galactosidase A, which catalyzes the cleavage of glycosphingolipids. While female carriers may have a milder and more variable clinical presentation when compared to hemizygous males, they experience renal and extrarenal symptoms, as well as morphologic changes, more often than previously reported. In fact, histologic features of Fabry disease have been identified in asymptomatic patients during workup as renal donors. This observation becomes relevant while determining when to initialize enzyme replacement therapy in female carriers without clinical symptoms of Fabry disease.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.