Clinical History:
The patient is a teenage male with past medical history end-stage renal dialysis and heart failure who presented with nausea/vomiting and diarrhea.
Which gene is classically mutated in congenital myopathy with cardiomyopathy and features seen in the provided images?
A. RYR1
B. TNNT1
C. C9orf72
D. TTN
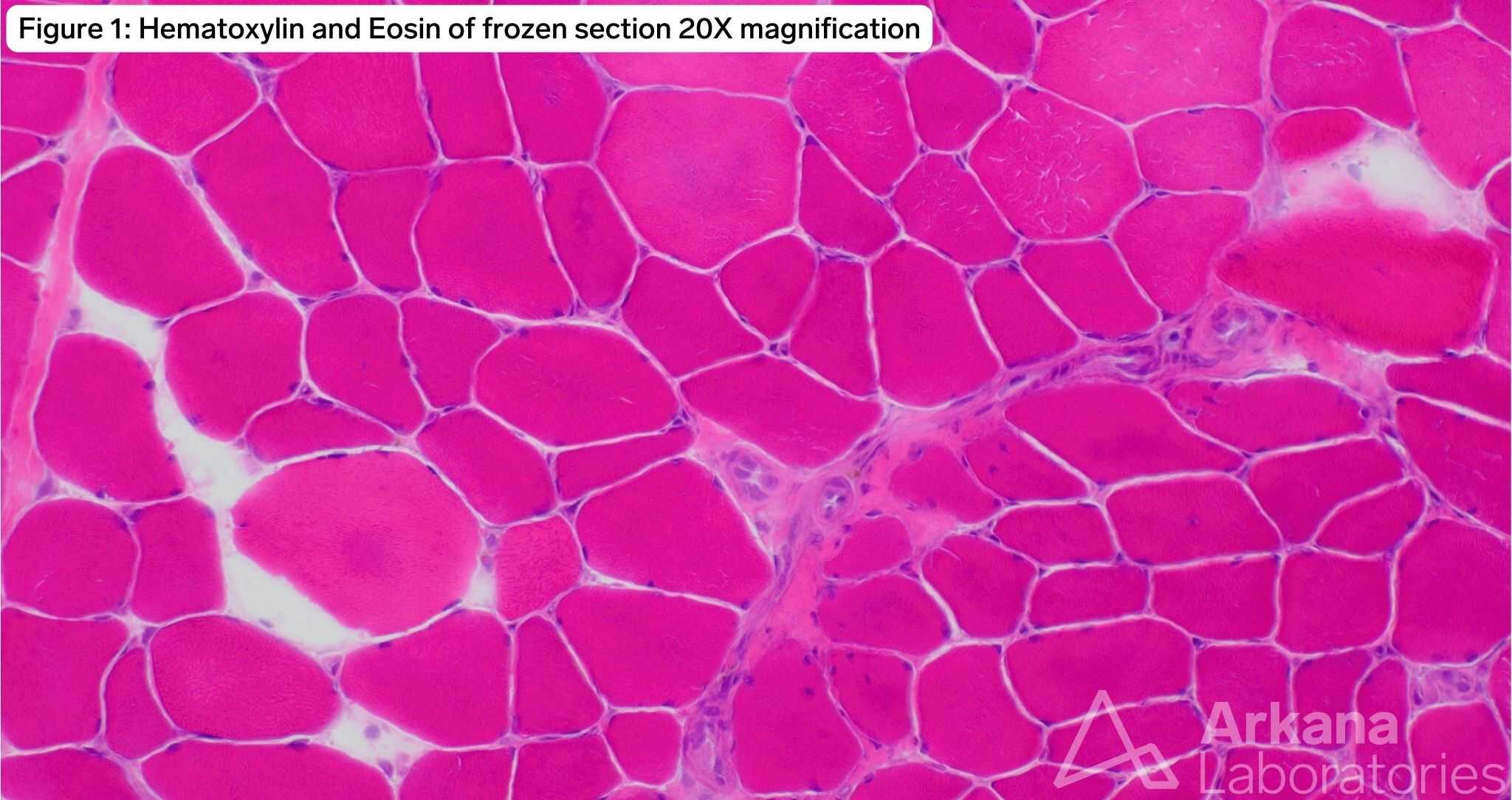
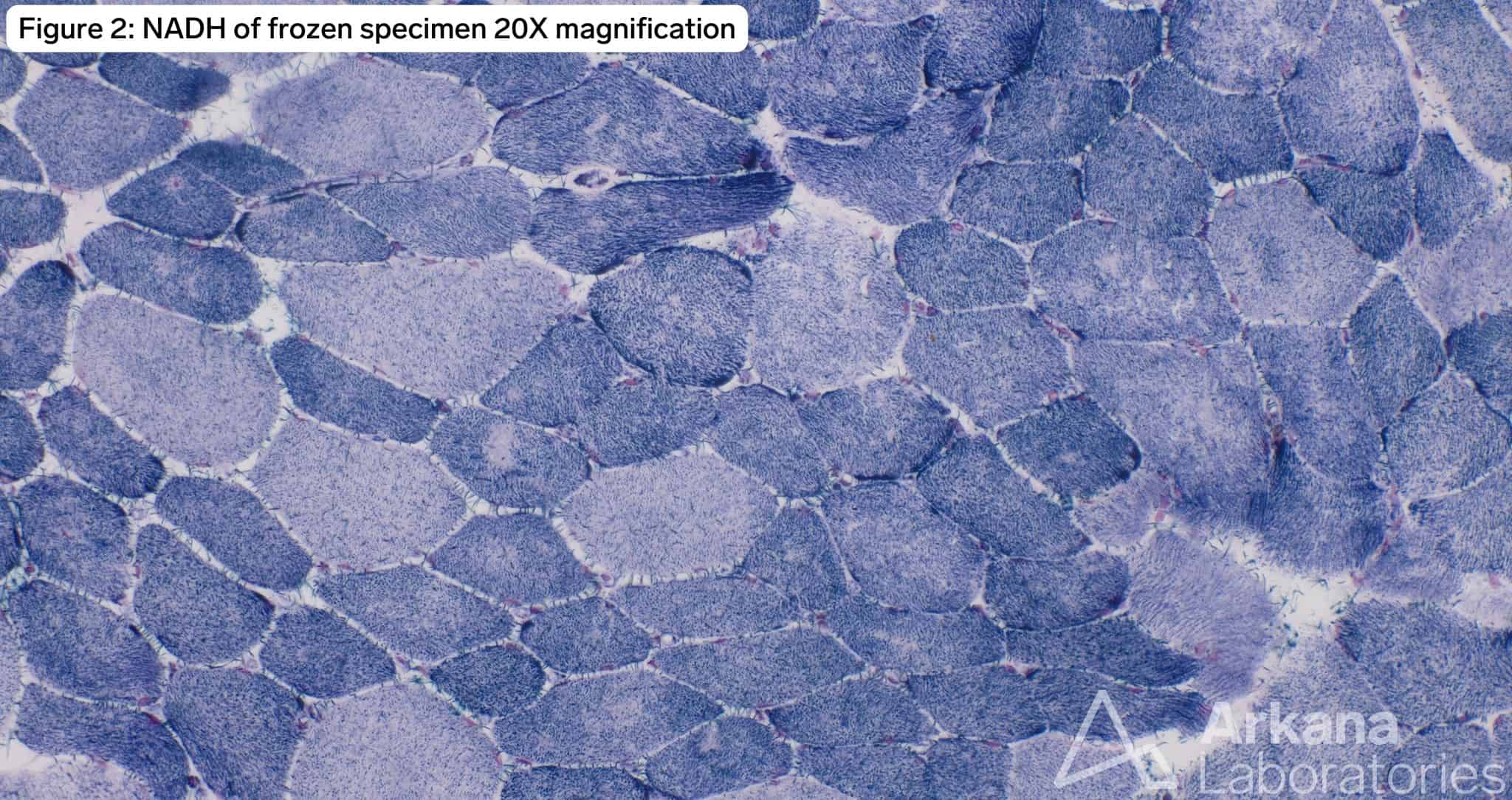
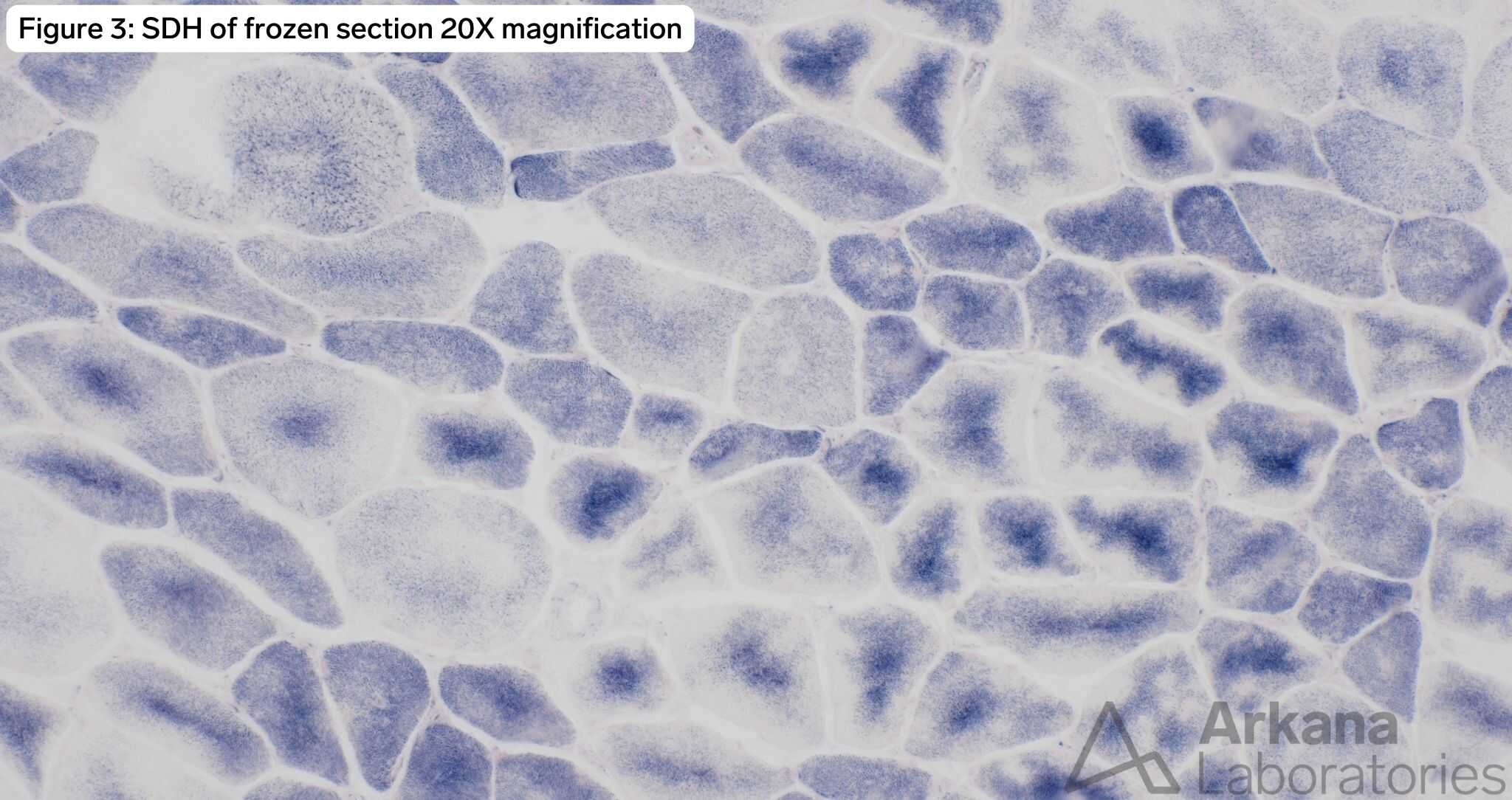
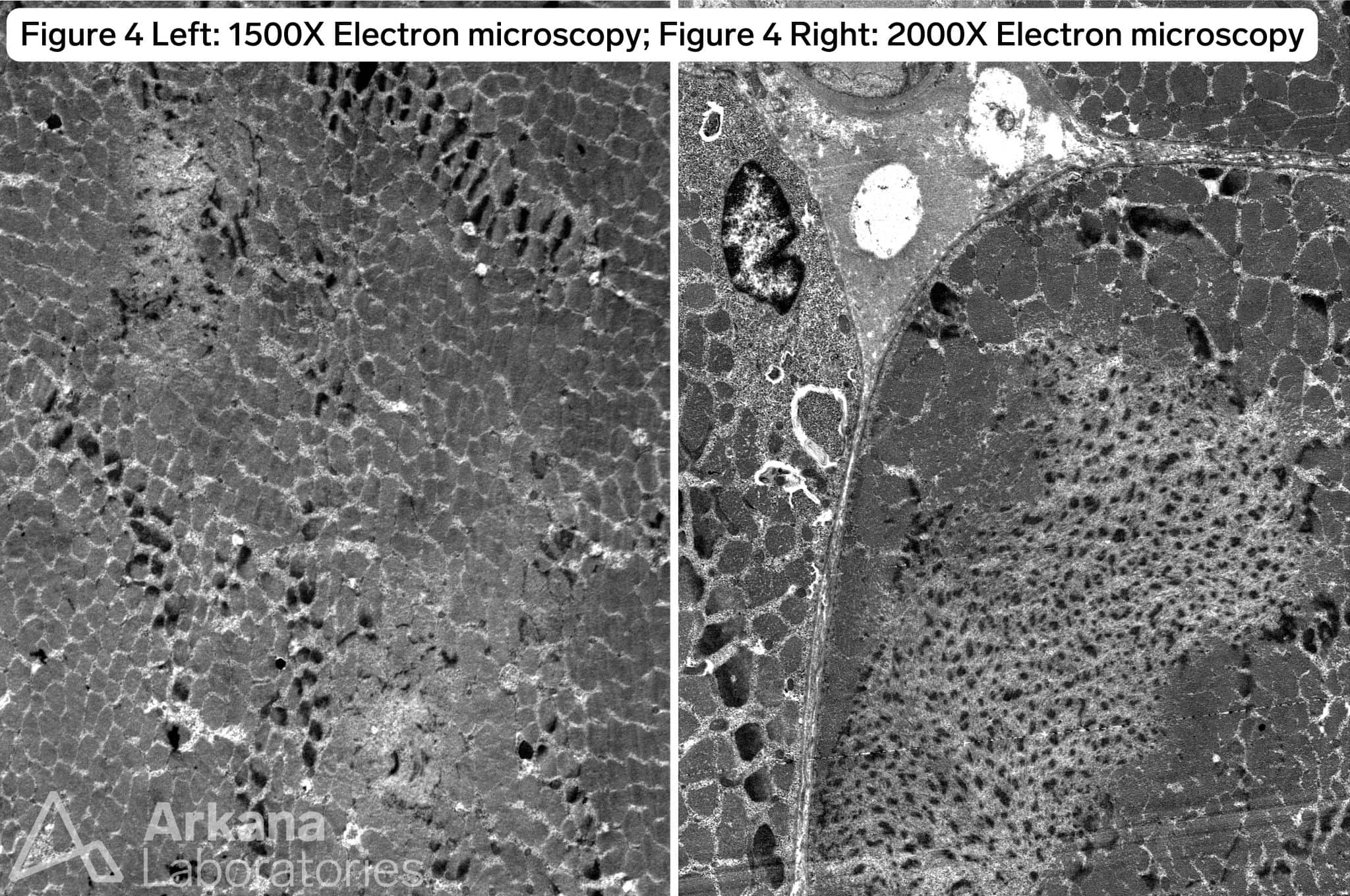
Answer:
D. TTN
The patient has a TTN c.60931C>T (p.Arg20311*) mutation known to be pathogenic for titin-related congenital myopathy. The TTN gene is one of the largest genes in the human genome with the most exons (364) and longest coding sequence of any human gene. TTN encodes for titin, the largest protein found in nature, which is expressed in both cardiac and skeletal muscle. The protein forms a complex with a second antiparallel titin molecule to form an elastic myofilament spanning the length of the sarcomere and acting like a spring. Figure 1 demonstrates frequent internalized nuclei focused in atrophic type I myofibers (fiber type confirmed by MHCf/s dual stain not pictured), as well as hypertrophied type II myofibers with both fiber types demonstrating pale central basophilia. Figures 2 and 3 highlight multiple areas within multiple myofibers with pale staining or complete loss of staining. Figure 4, the ultrastructural examination of these pale staining areas demonstrates myofibrillar disruption, often multiple areas within a single myofiber. Together these findings are consistent with multiple mini cores. Combining these features with the history of cardiomyopathy raises strong concern for titin-related congenital myopathy, when combined with the genetic information the diagnosis is confirmed.
The RYR1 gene codes for the skeletal muscle ryanodine receptor on chromosome 19q. Mutations are seen in central core disease which can present with multiple minicores, however cardiomyopathy is not a feature of RYR1 driven central core disease due to the gene product being a skeletal muscle specific receptor.
The TNNT1 gene codes for skeletal troponin T. While mutations in TNNT1 lead to congenital myopathy the main histologic features is that of rods, though cores may also be seen. As well, while patients with TNNT1 mutations have been reported to present with dilated cardiomyopathy it is a less common feature than is seen in cases with pathogenic TTN mutations.
C9orf72 is mutated in familial ALS. Core areas demonstrate significant histologic overlap with target and targetoid areas in dennervated myofibers. However, the cores in this case do not demonstrate the intermediate vs central zone on electron microscopy or rim of darker NADH staining surrounding the pale center.
Reference(s) / Additional Reading:
- Titin-related congenital myopathy
- Oates EC, Jones KJ, Donkervoort S, et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann Neurol. 2018;83(6):1105-1124. doi:10.1002/ana.25241
- TNNT1 nemaline myopathy with cardiomyopathy
- Streff H, Bi W, Colón AG, Adesina AM, Miyake CY, Lalani SR. Amish nemaline myopathy and dilated cardiomyopathy caused by a homozygous contiguous gene deletion of TNNT1 and TNNI3 in a Mennonite child. Eur J Med Genet. 2019;62(11):103567. doi:10.1016/j.ejmg.2018.11.001
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.