
We received the biopsy from a 25-year-old female who presented for evaluation of nephrotic range proteinuria. Lab evaluation reveals a creatinine of 0.55 and 24-hour urine protein is 3.6 g. Serologies were negative or normal for ANA, hepatitis B, hepatitis C, HIV, and complement levels. There was no history of hypertension or diabetes.
A biopsy was performed to evaluate the source of proteinuria.
Two cores of renal tissue were sampled on light microscopy evaluation. They consisted mostly of medulla, and only two glomeruli were seen in multiple sections.
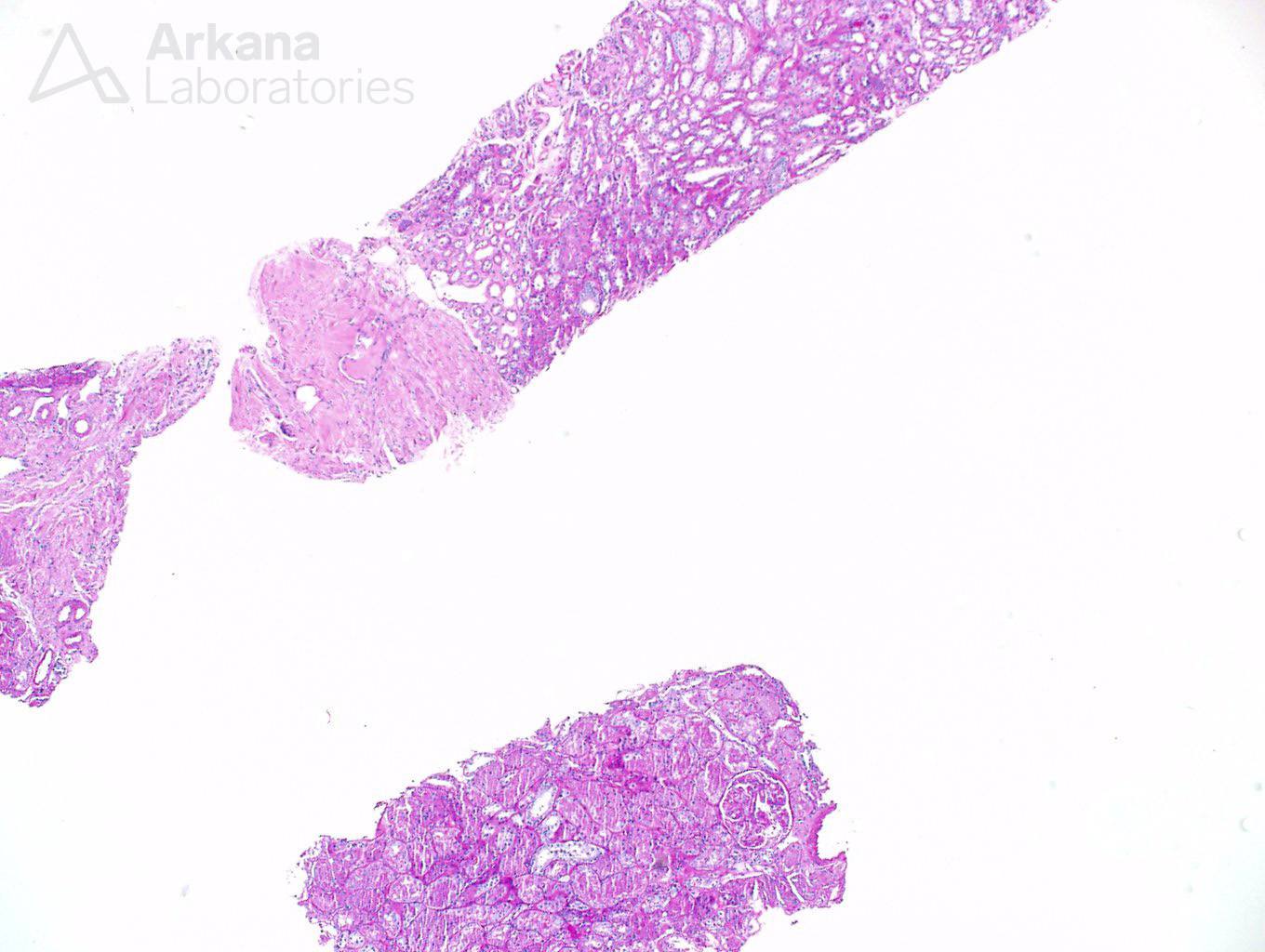
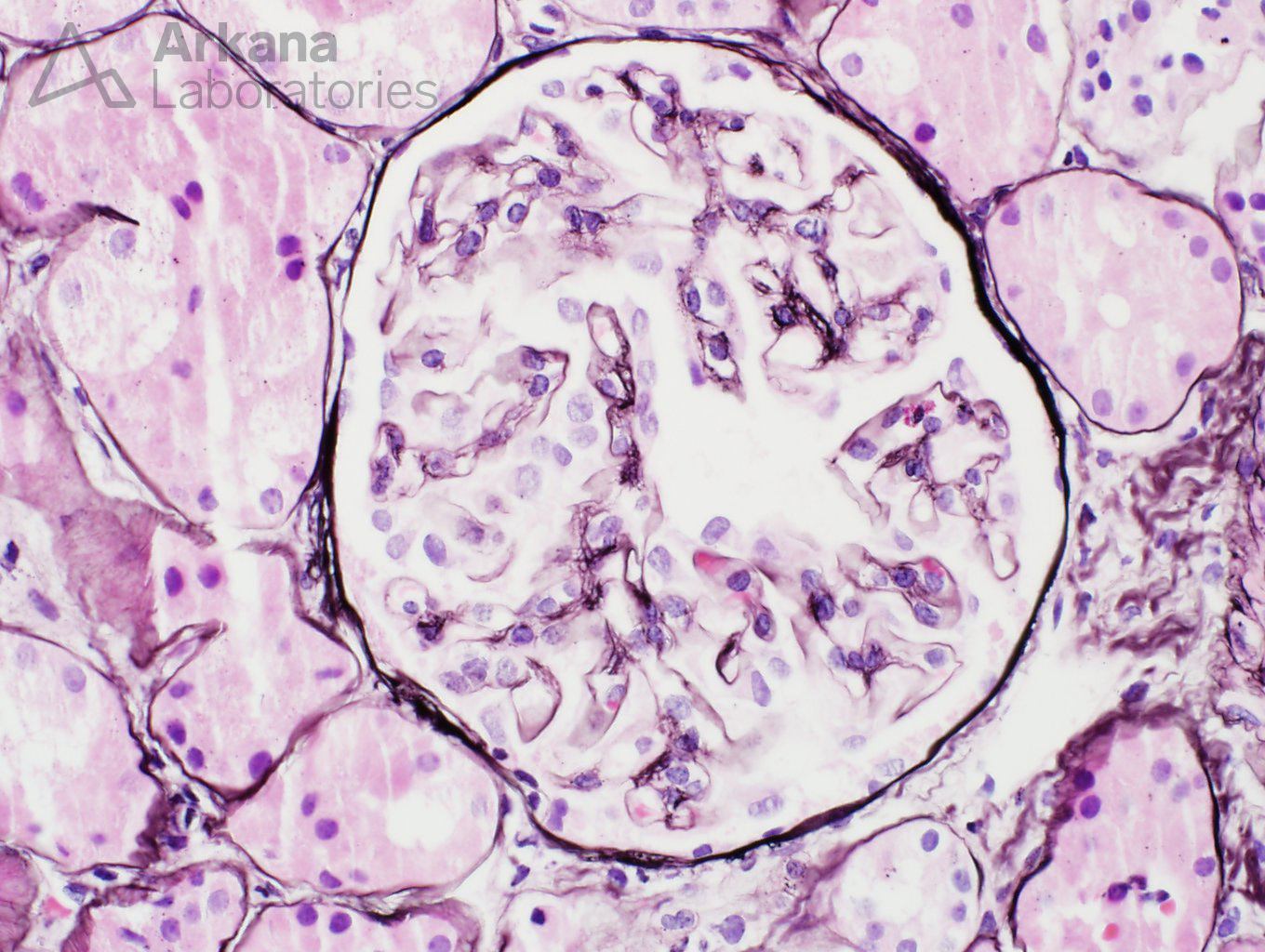
The glomeruli have capillary loops with attenuated contours. The capillary loops appeared delicate. But no other changes were promptly noticeable. Features of the usual culprits for nephrotic syndrome- membranous nephropathy and FSGS – were not readily identifiable on this sample.
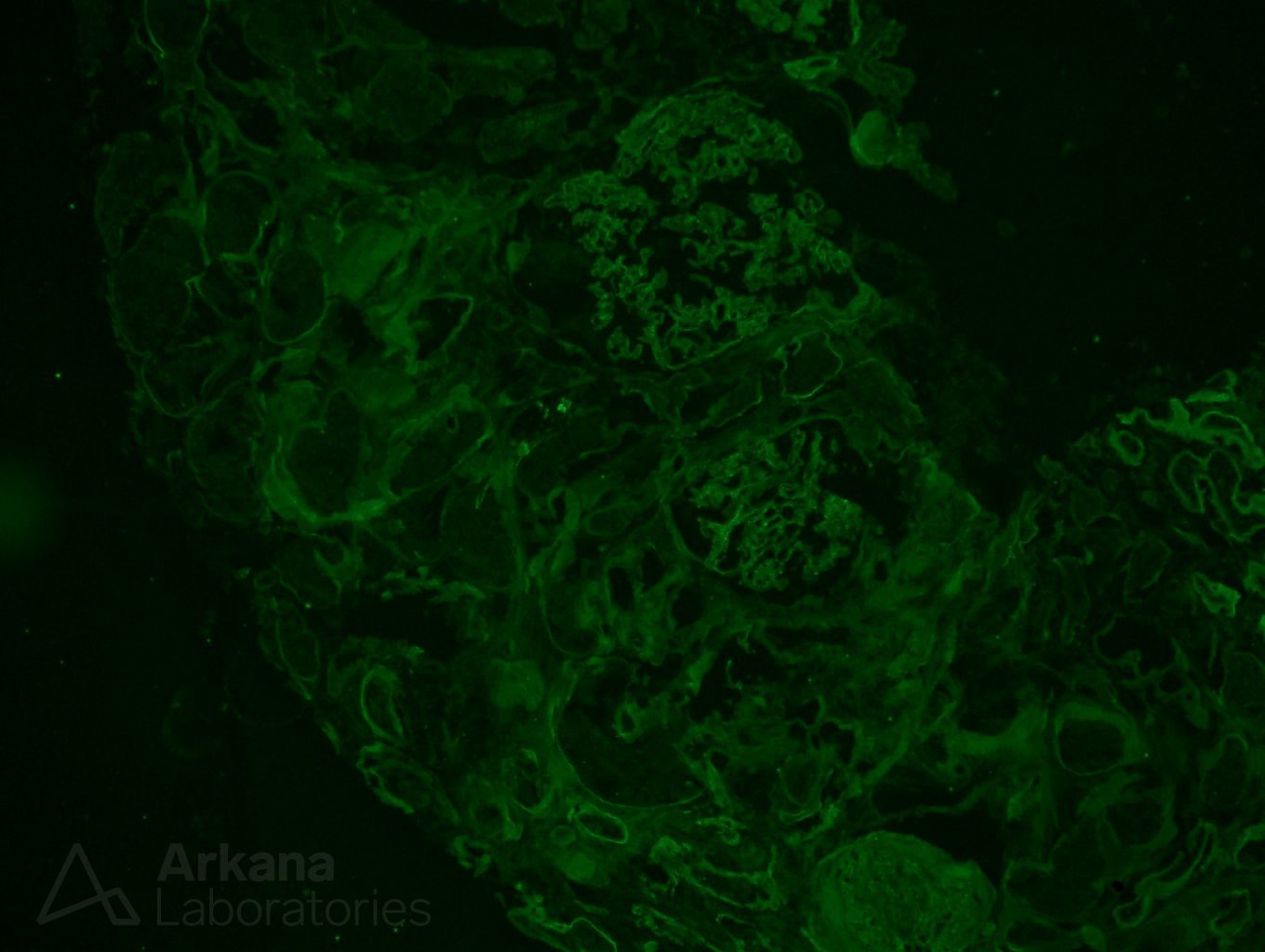
Immunofluorescence studies were negative for all immunoglobulins, complement factors and light chains, in the single glomerulus present on these sections. And the tissue submitted for electron microscopy did not contain glomeruli.
At this point, all seemed settled, or from another perspective, all seemed lost. Our pathologist was unsatisfied with the small sample, which by all means seemed quite inadequate for a proper diagnosis. The possibility of an immune complex mediated disease was improbable, due to negative immunofluorescence, however a definitive diagnosis could not be rendered. Still, in the spirit of trying further, and leaving no stone unturned, our pathologist decided to deeper the light microscopy sections, in the hope of shading some light into the case. Maybe luck would be on our side?
Multiple deeper sections were examined and the number of glomeruli present in the tissue increased from two to a whooping three. Not much gained after all. The glomeruli did not bring any new information with them. But a seemingly insignificant finding was noted in the first deeper sections- a number of interstitial foamy cells were present in the cortical region.
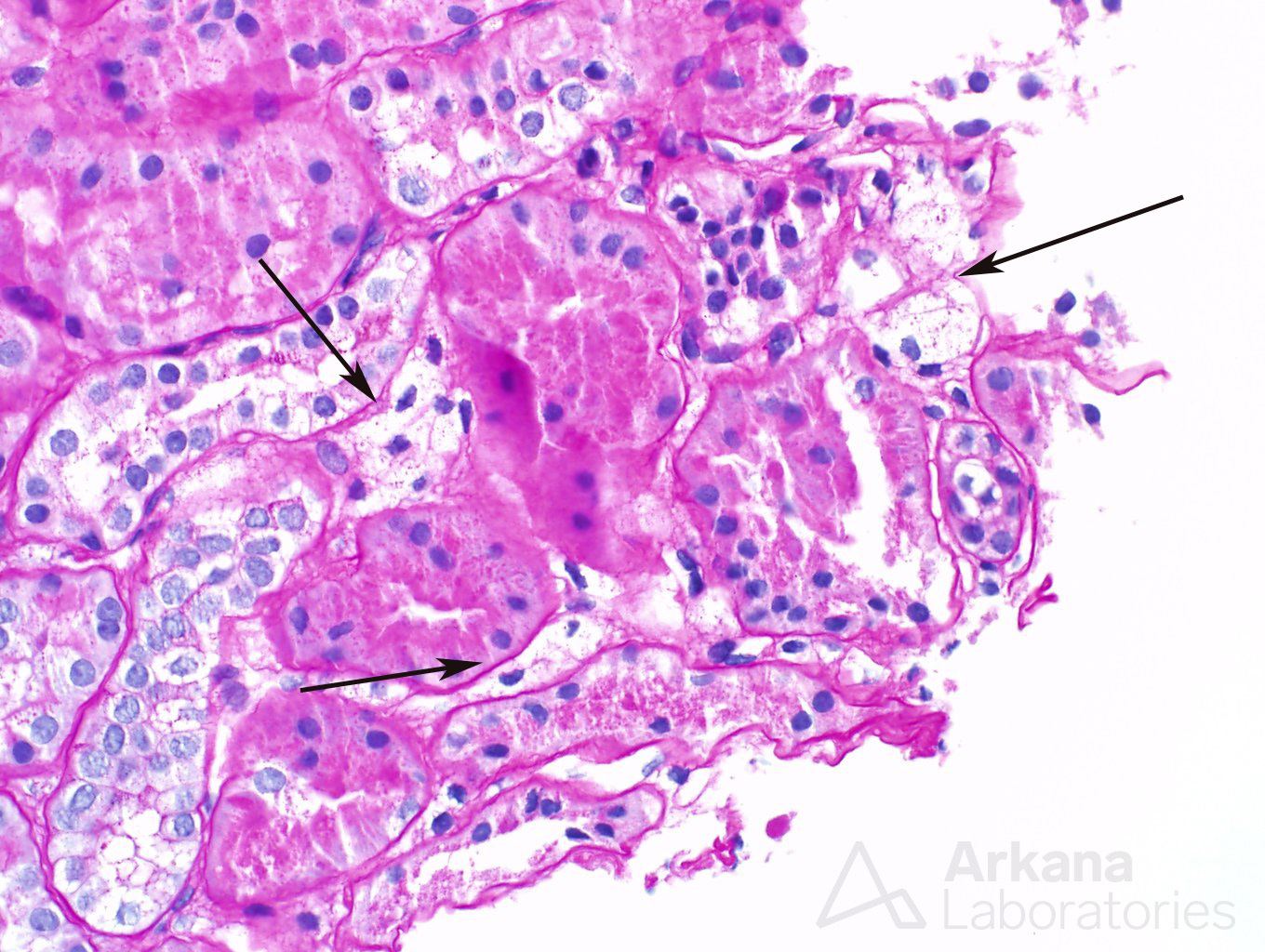
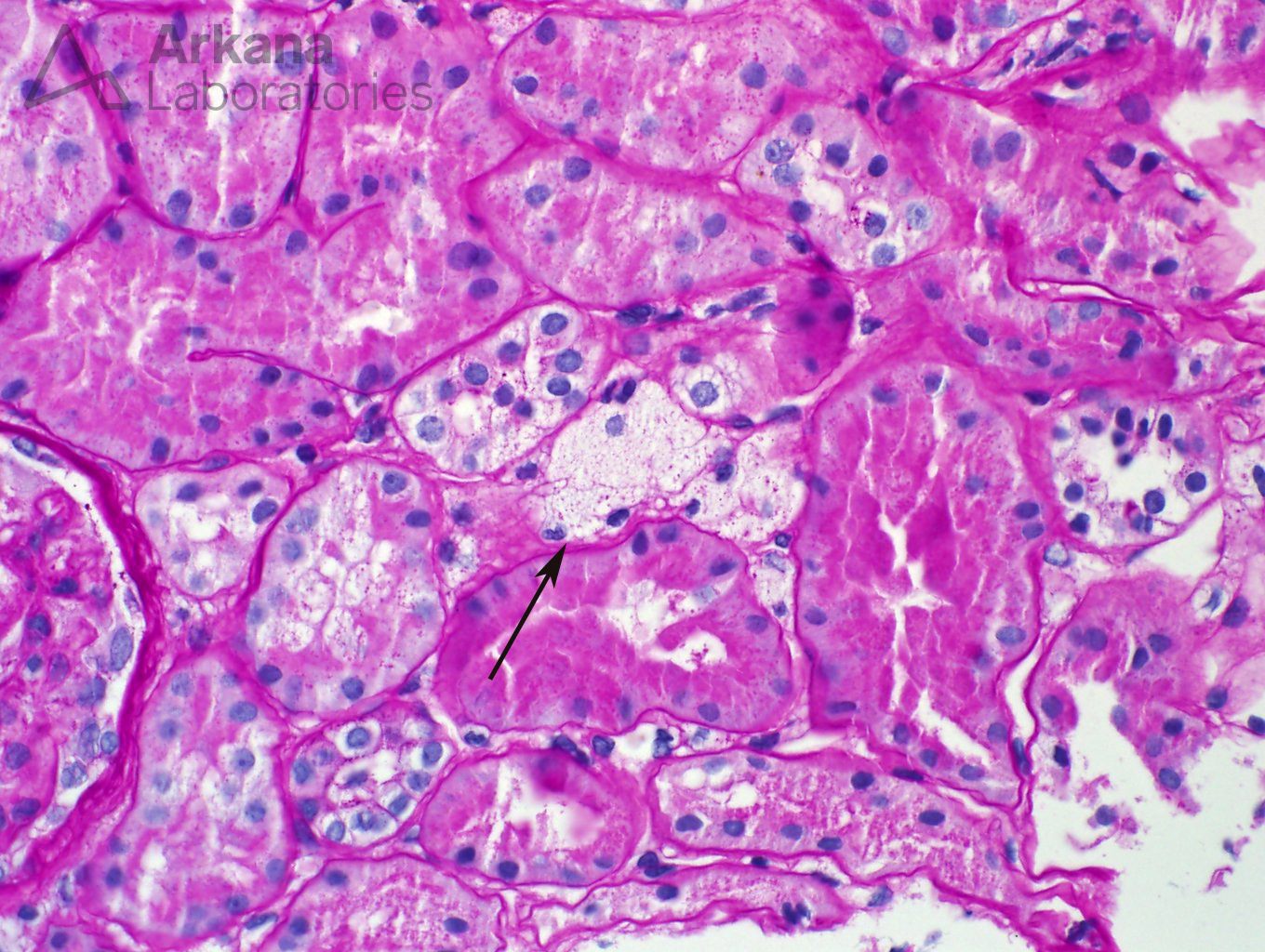
Foamy cells are a finding that may be quite non-specific in patients with proteinuria. One can find them at any single entity that is associated with nephrotic syndrome. But the presence of foamy cells also brings a more interesting diagnosis into the differential (and a dear question of pathology and nephrology boards): Alport syndrome.
Given that the patient was young, our pathologist decided to pursue that improbable diagnosis to the end. An immunofluorescence stains for Alpha 2 and 5 chains of collagen type IV was ordered. Absence of staining for alpha 5 is consistent with a diagnosis of Alport disease. Observe below a normal control case where alpha 2 (red stain) and alpha 5 (green stain) are seen along glomerular basement membranes. To the side on the control case, one can see the same immunofluorescence applied to our patient.
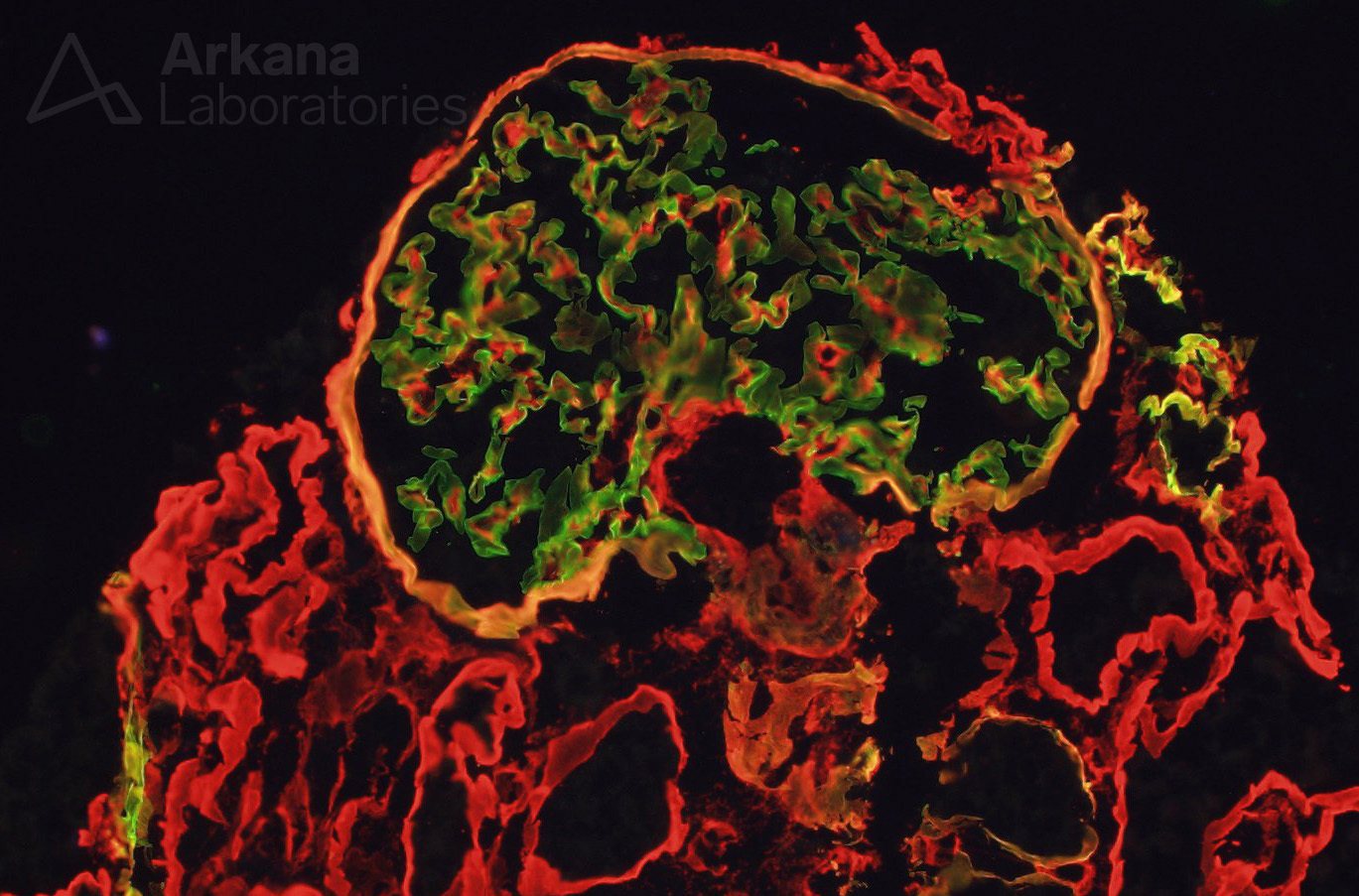
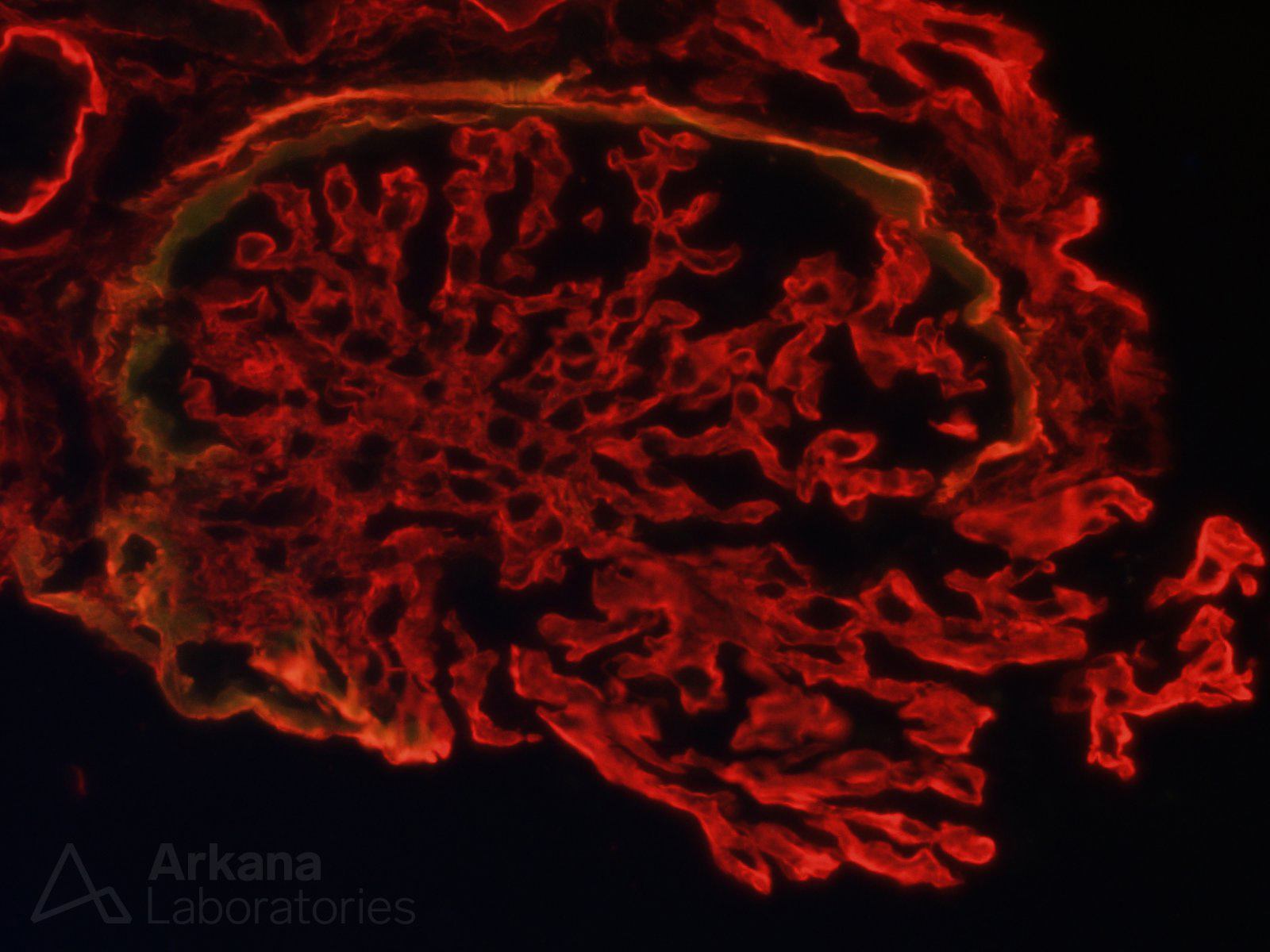
These results were astounding! The immunofluorescence stain for alpha 5 chain of collagen type IV was absent in our patient. The foamy cells in this case were indeed likely related to Alport syndrome. That was a phenomenal result, but more digging was going on at the same time that the immunofluorescence studies were cooking: multiple additional deeper sections were examined on the material submitted for electron microscopy. And one intact glomerulus was found on deeper sections #7 (lucky one).
Electron microscopy revealed altered basement membranes with findings including lamellation, and irregularity, and extremely thin GBMs in certain areas
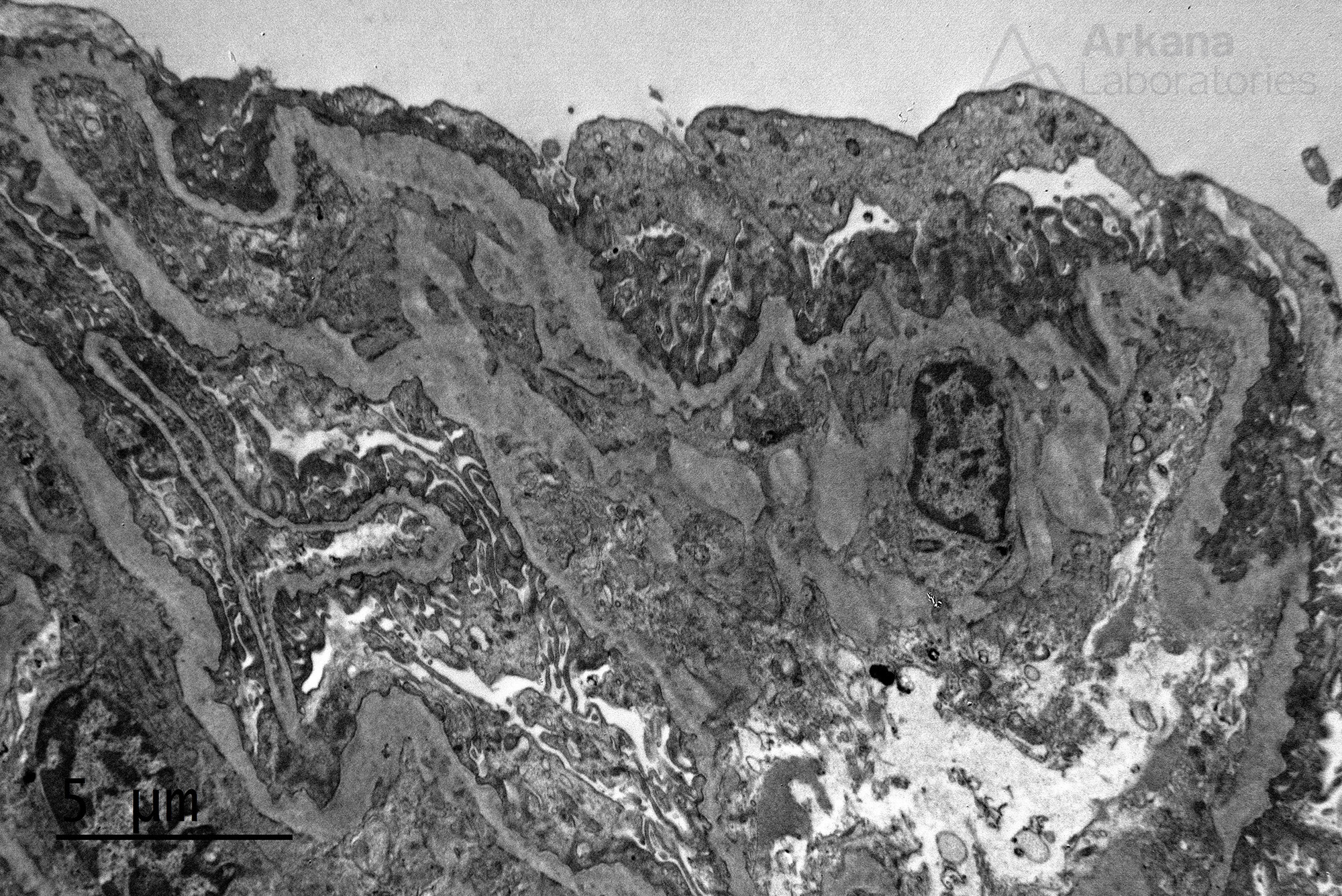
Altogether, the findings on the biopsy were diagnostic of Alport syndrome. And from what was inadequately sampled we landed on a pretty rare diagnosis. We dug deeper and we went far, after all: “Only those who will risk going too far can possibly find out how far one can go” (T.S.Eliot).
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.