A 15-year-old boy had a kidney biopsy because of the persistent nephrotic syndrome. His medical history is significant for resolved intermittent asthma. In late September 2016, he developed a viral syndrome with a runny nose, congestion, sore throat, vomiting, and diarrhea. Over the next one to two weeks, he developed generalized edema, most prominent in the lower extremities and belly. He was diagnosed presumptively with acute glomerulonephritis and treated accordingly. His condition continued to worsen with the persistent nephrotic syndrome and acute hypertension. Laboratory investigations show a serum creatinine of 1.35 mg/dL. Serum albumin is 1.9 g/dL. Urinalysis shows 3+ proteinuria, 1-2 RBC/hpf and 2-3 hyaline casts/hpf. Serological studies are negative for ANA, anti-GBM, ANCA, MPO, PR3 and ASO. There is hypocomplementemia with C3 levels of 51 mg/dL and C4 of 13.6 mg/dL.
The images are characteristic findings of which of the following:
A) Collapsing glomerulopathy
B) Dense deposit disease
C) Lupus nephritis
D) Infection associated crescentic glomerulonephritis
E) C3 glomerulopathy
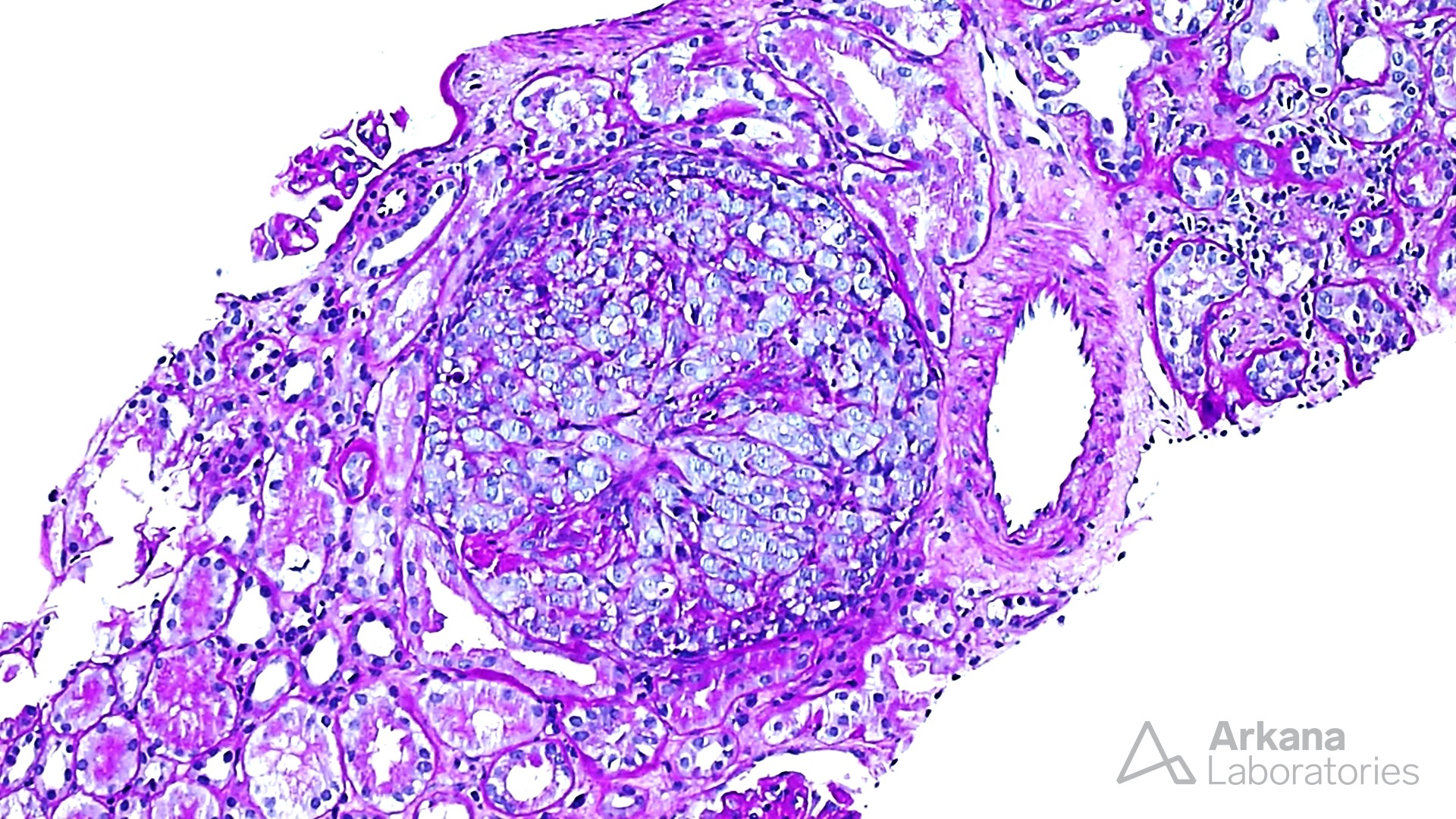
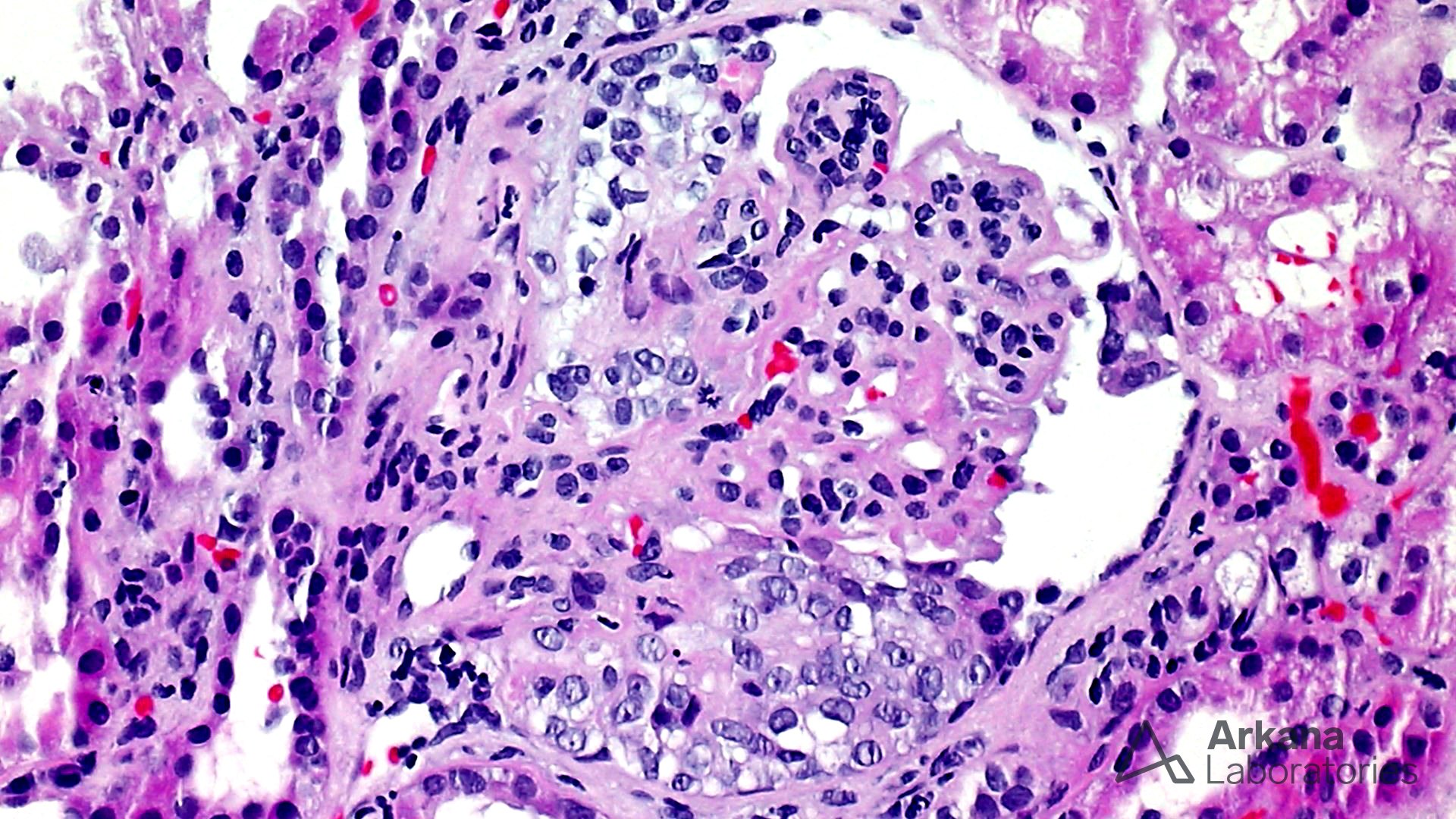
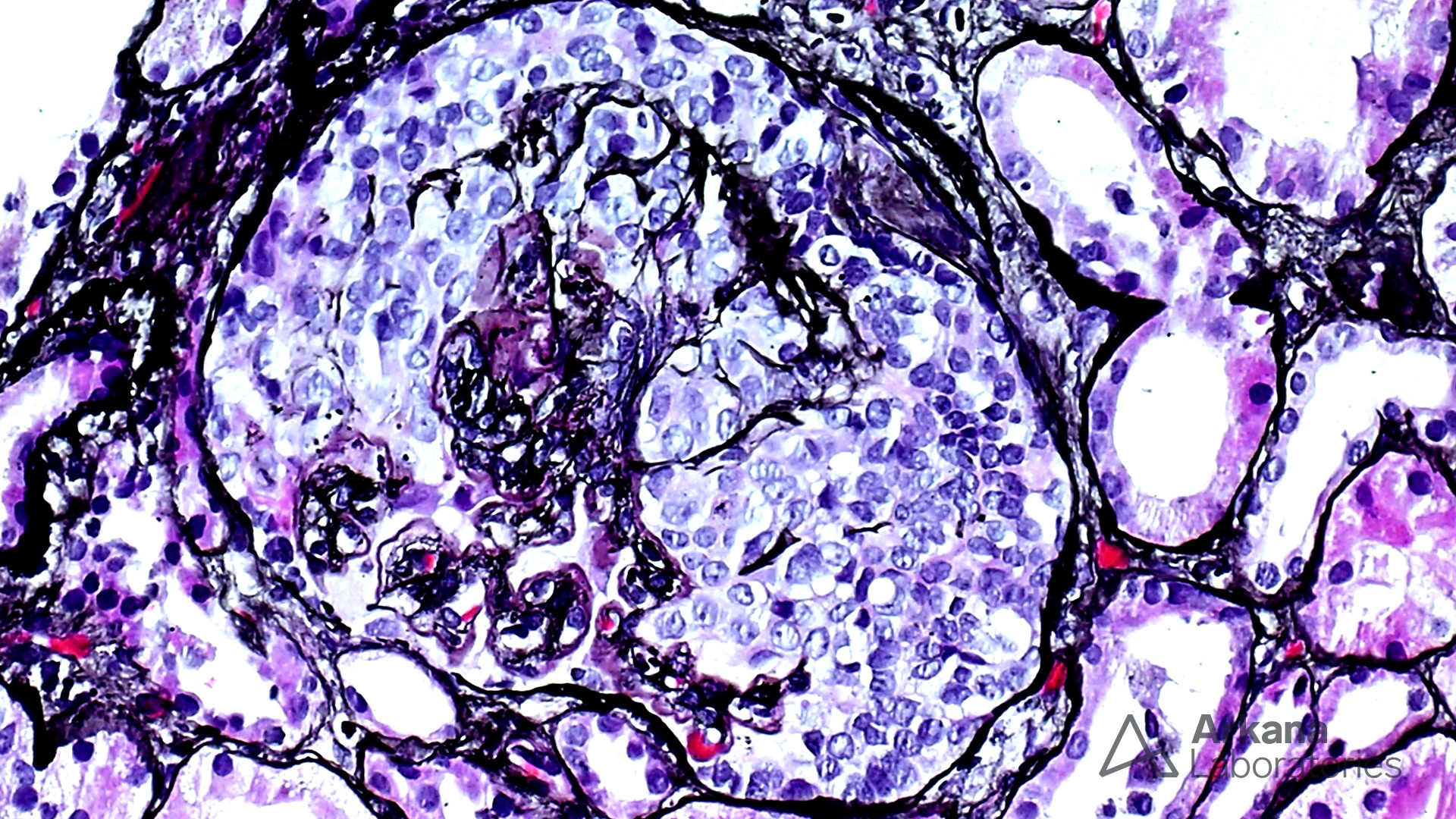
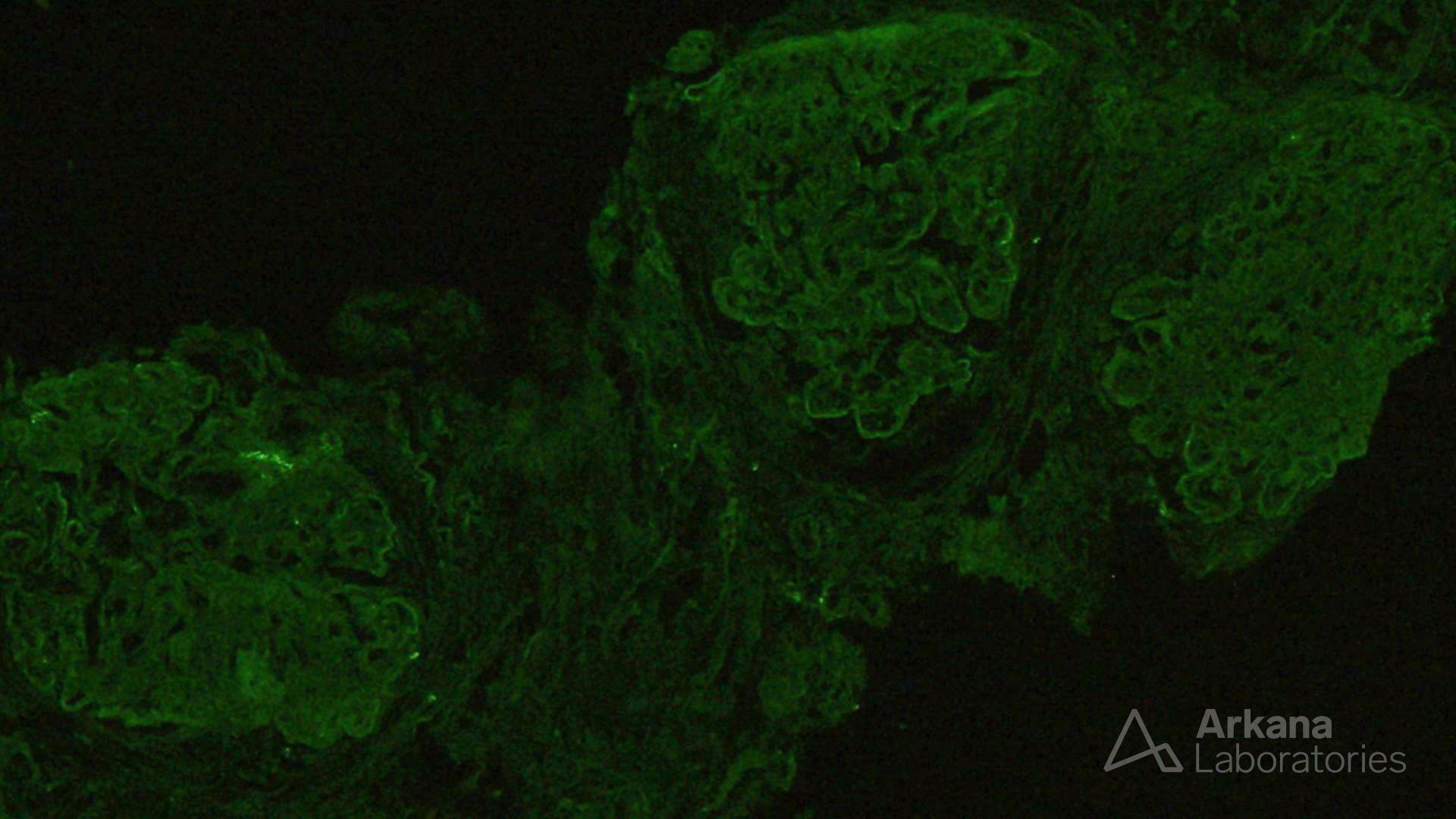
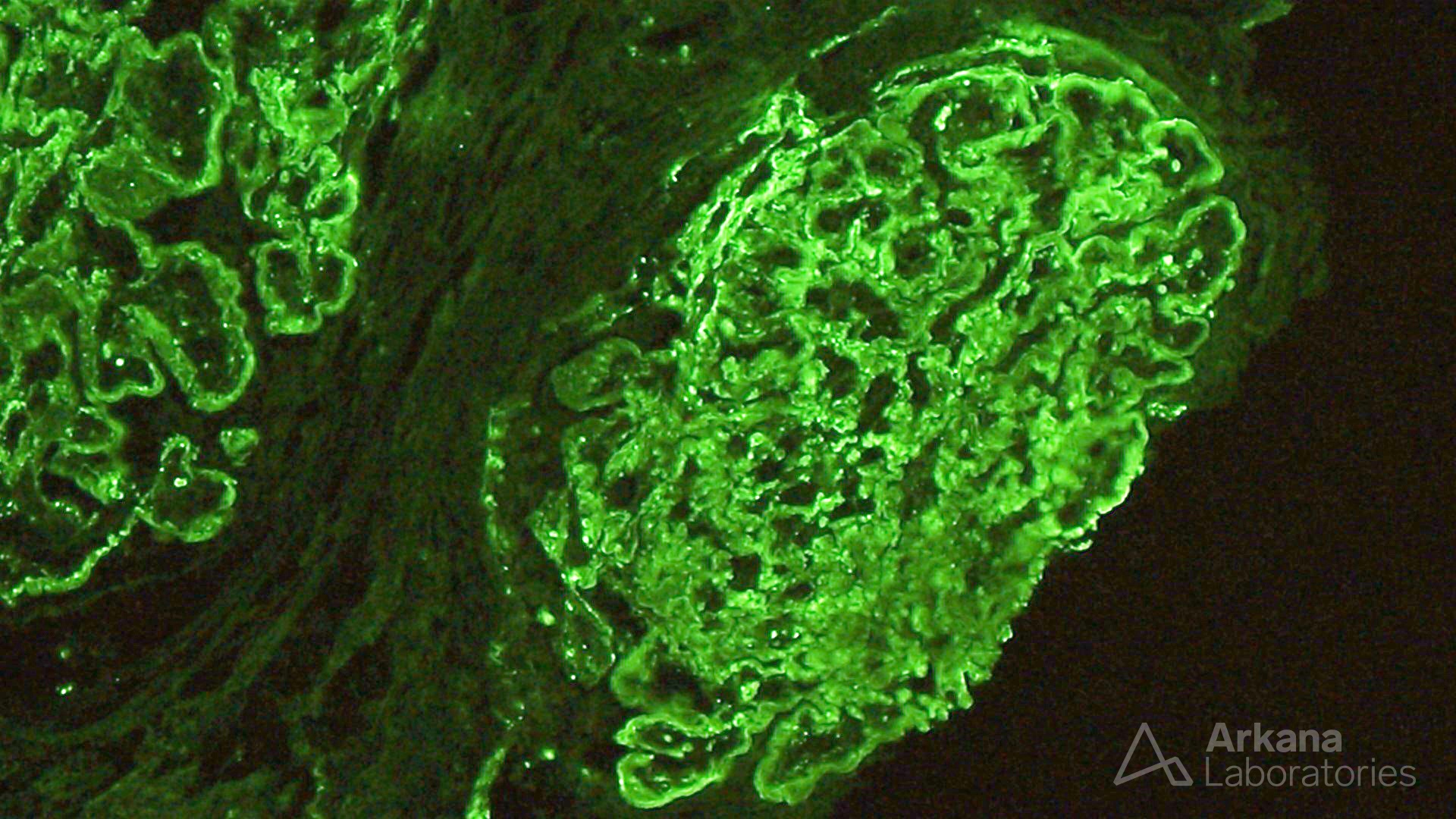
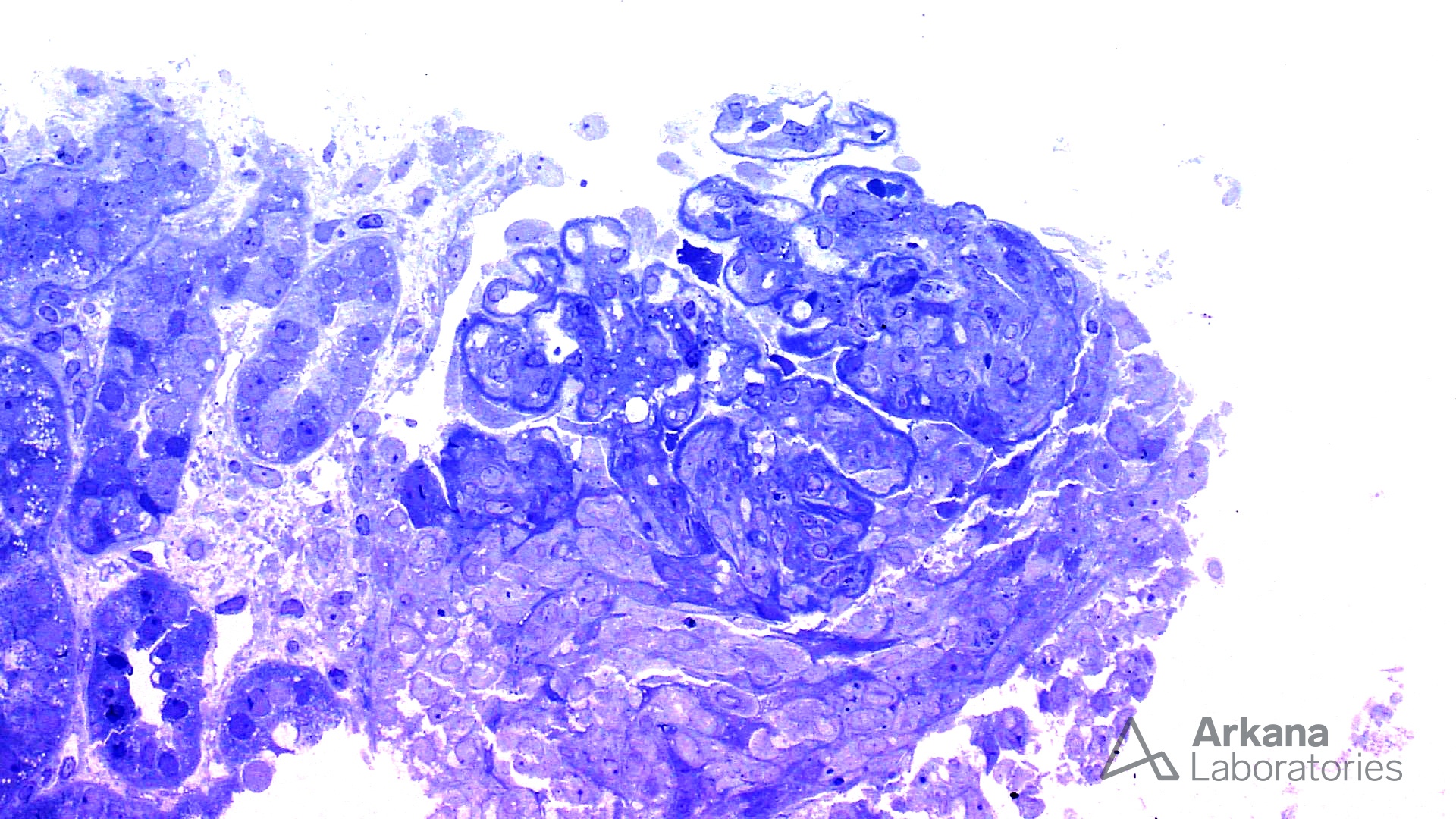
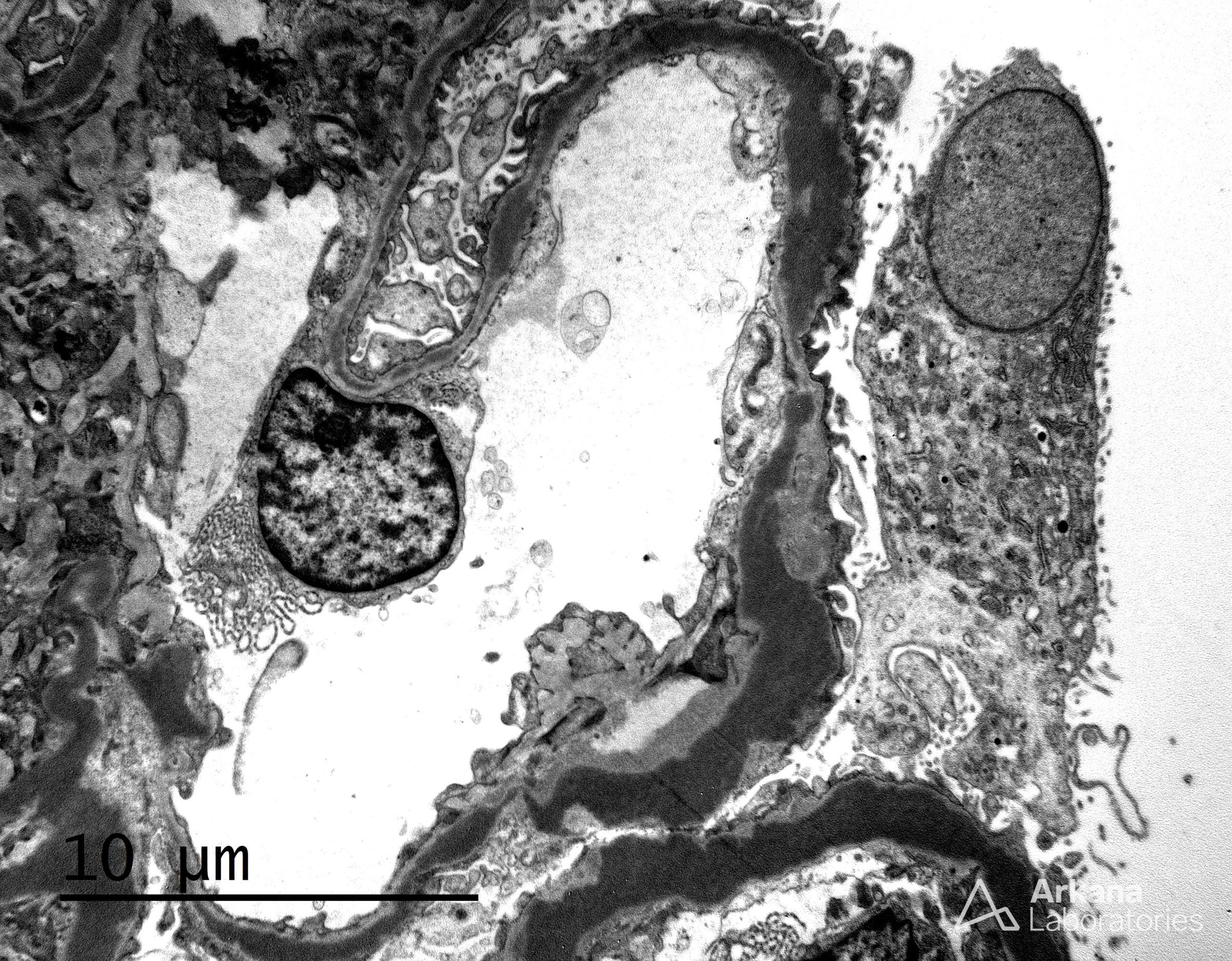
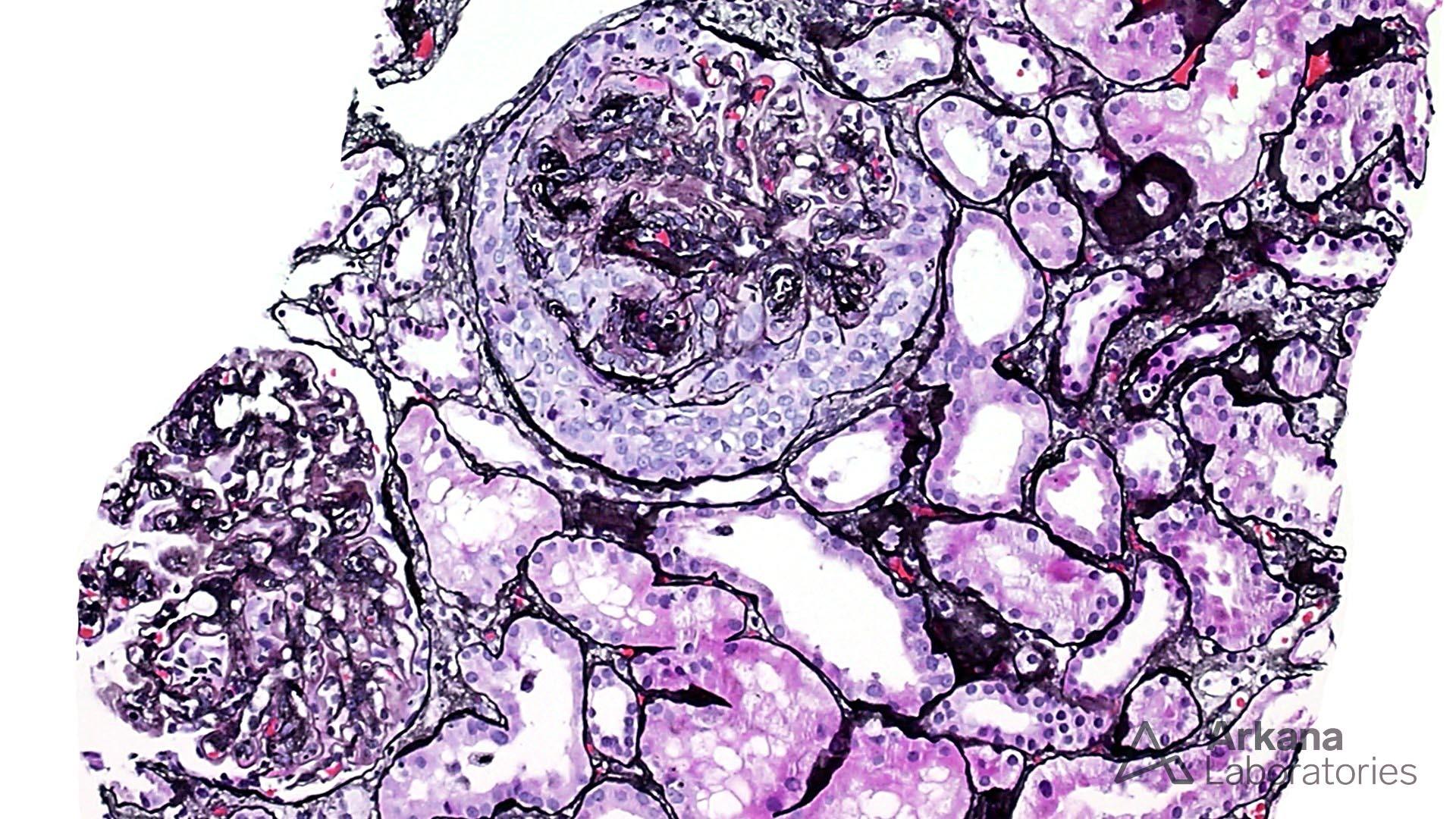
The features shown are those of dense deposit disease (DDD) (choice B). There is a proliferative crescentic glomerulonephritis, involving almost all of the glomeruli sampled. However, the most characteristic findings are predominantly C3 staining in the glomeruli with occasional mesangial rings, along with electron microscopy showing linearized and sausage-shaped osmiophilic deposits within glomerular basement membranes.
Dense deposit disease is a C3-related glomerulopathy characterized by the broad, linear and extremely electron dense deposits with the GBM, mesangium, Bowman’s capsule, and tubular basement membranes. The hyperdense osmiophilic deposits are pathognomonic, and hence the term dense deposit disease.
DDD is thought to be due to chronic activation of the alternative complement pathway. This may be due to the formation of autoantibodies to components of the pathway that contribute to inactivation of C3, such as C3NeF (C3 nephritic factor), complement factor H, factor B or C3. Autoantibodies to C3NeF are present in 100% of children and 40% adults. Genetic factors such as mutations in Factor H are implicated in 15 – 20% cases. Patients often present with proteinuria (95%; 60% nephrotic range), hematuria (90%) and renal insufficiency (50%). ~80% patients have low serum C3 levels. ~50% patients develop end-stage renal disease in 10-15 years. Unfortunately, the disease tends to recur in almost all allografts, and ~50% allografts fail within 3 years.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.