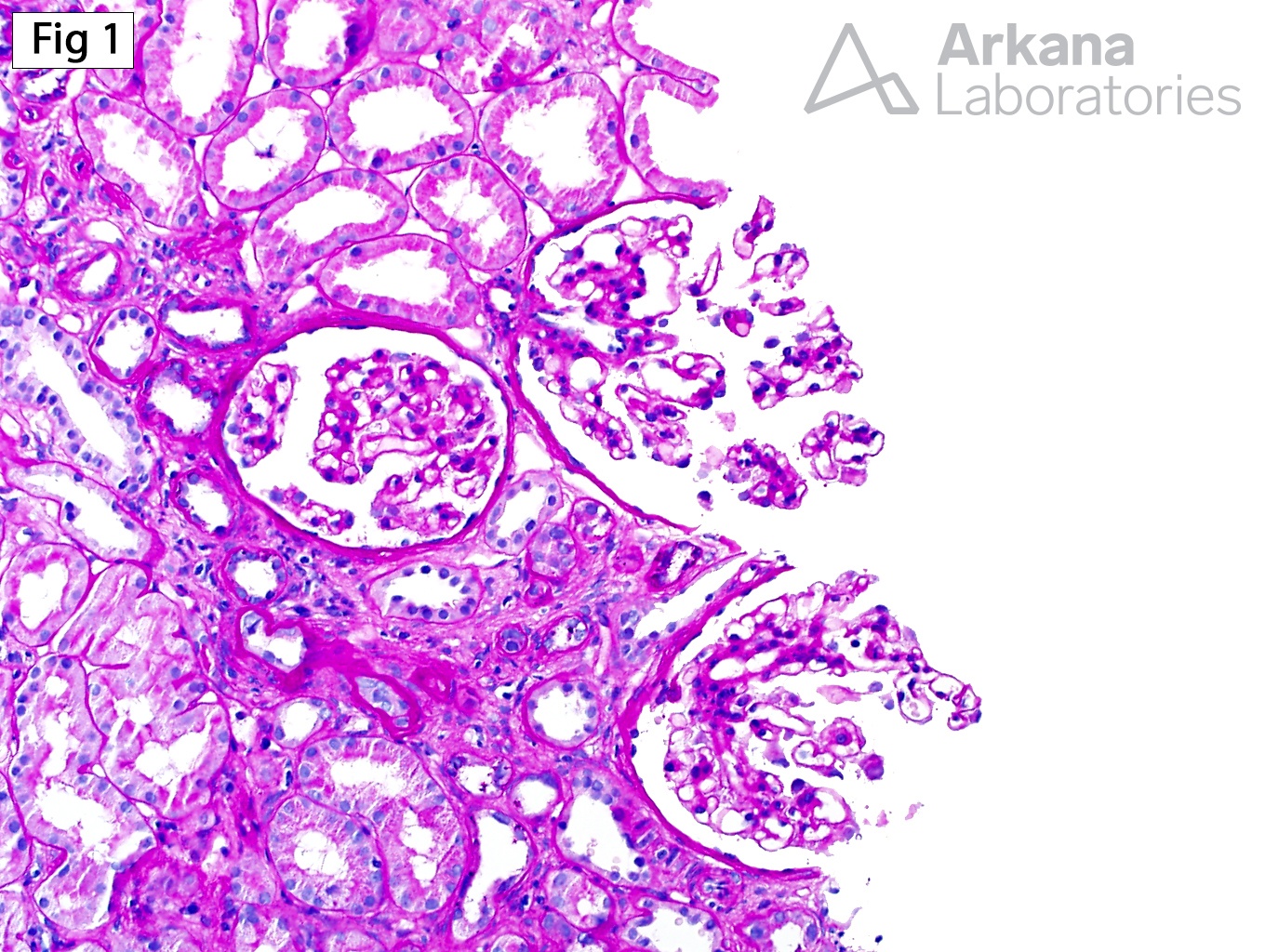
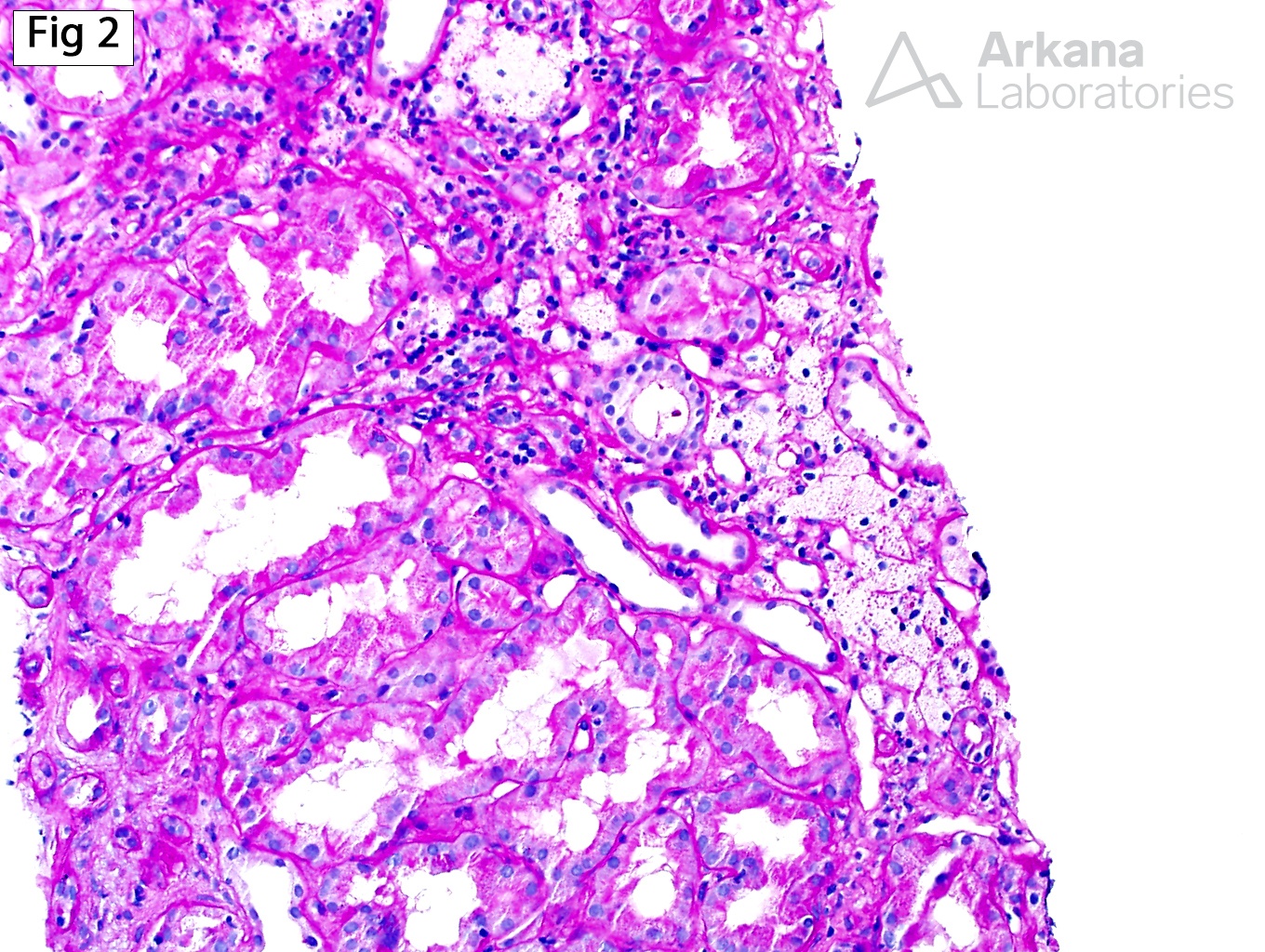
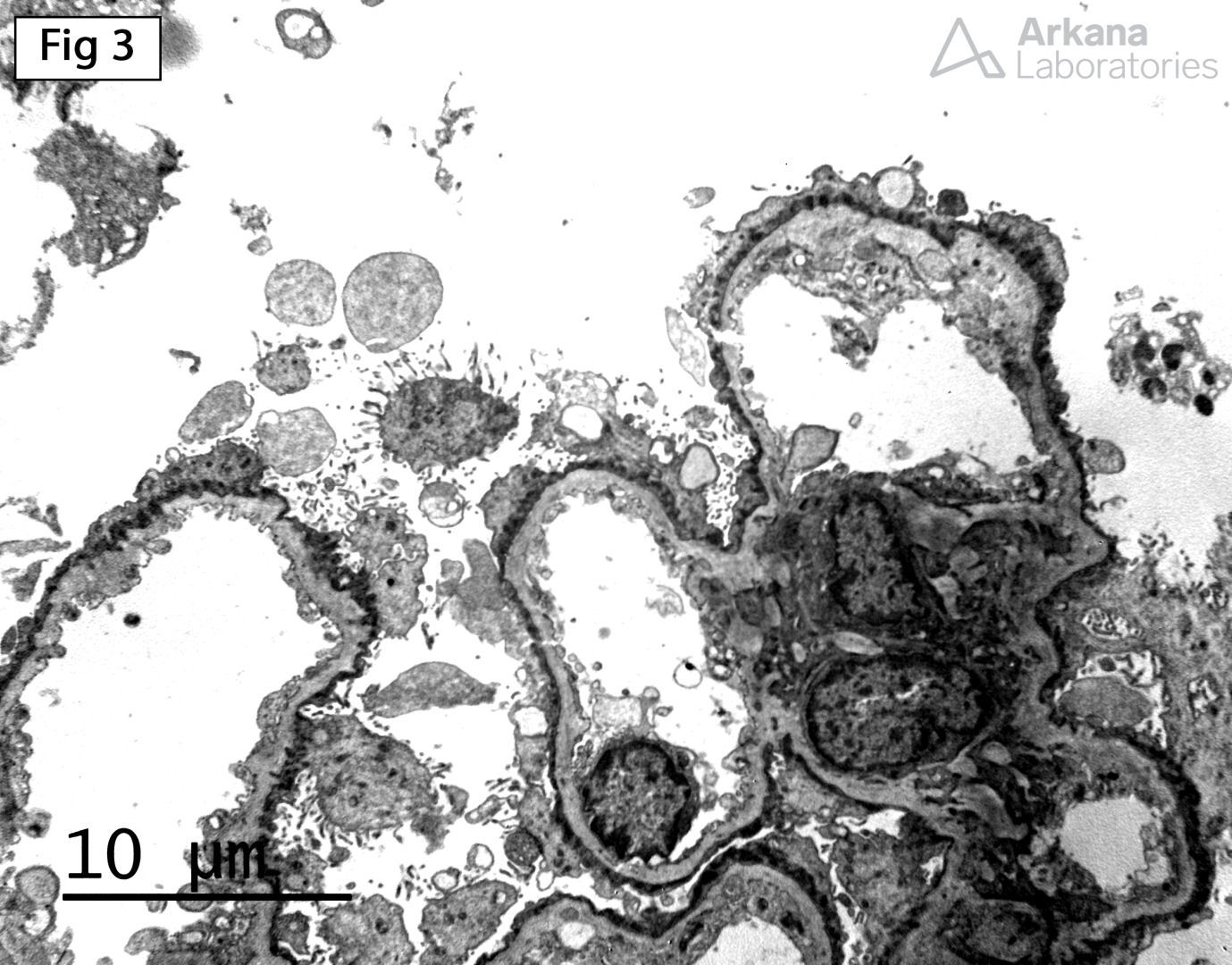
This biopsy from a 22-year-old Hispanic male with a history of hematuria, subnephrotic proteinuria, and chronic kidney disease shows essentially unremarkable glomeruli (Fig 1) with moderate interstitial fibrosis and tubular atrophy, and frequent interstitial foam cells (Fig 2). Furthermore, electron microscopy shows irregular glomerular basement membranes (GBM) that vary between thick and thin, along with segmental lamellation and diffuse epithelial foot process effacement (Fig 3). These findings, along with the clinical history are consistent with Alport syndrome. Alport syndrome is a spectrum of diseases affecting the synthesis of type IV collagen, which is the main component of the glomerular basement membranes. Although the clinical presentation varies slightly based on the underlying genetic abnormality, patients with Alport syndrome usually present in childhood with hematuria, proteinuria, progressive renal insufficiency, sensorineural hearing loss and lenticonus. Approximately 80 – 85% of patients show X-linked dominant inheritance due to mutations in the COL4A5 gene, located on chromosome Xq26-48. Additionally, patients may show mutations in the COL4A3 and COL4A4, with autosomal recessive (10-15% of patients) or autosomal dominant (5% of patients) inheritance. While X-linked and autosomal recessive forms of Alport syndrome have a similar clinical presentation, the autosomal recessive form may be suspected clinically by the presence of severe disease in young females, consanguinity in the family or presence of hematuria in the father of a male patient. On the other hand, autosomal dominant Alport syndrome typically has a milder clinical presentation than the X-linked or autosomal recessive forms, with only approximately 25% of patients progressing to ESRD, and those who do so are usually over 40 or 50 years of age.
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.