
Monday
Rediscovery of Complement
Mesangial C3 deposition only within the glomerulus has been reported since 1980. Only recognized recently as a distinct entity when CFHR5 nephropathy was reported. CFHR5 mutation is speculated to represent a founder mutation which can be traced back to the Troodos mountains of Cyprus. https://www.ncbi.nlm.nih.gov/pubmed/21566112

1 in 6500 people in Cyrus carry a mutation in CFHR5 (duplication of exons 2 and 3) and inheritance is autosomal dominant. C3 and C4 complement levels are normal. 80% of males with CFHR5 nephropathy progress to ESRD while only 20% of females progress to ESRD. https://www.ncbi.nlm.nih.gov/pubmed/22065842
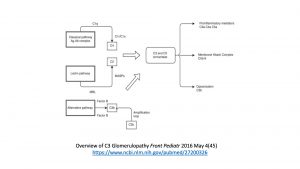
CFHR5 nephropathy is a subset of C3 glomerulonephritis. CFHR5 nephropathy opened pandoras box to look at other proteins which regulate C3 & C5 convertases.
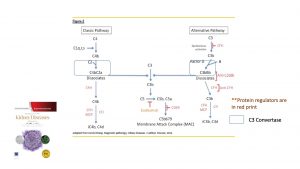
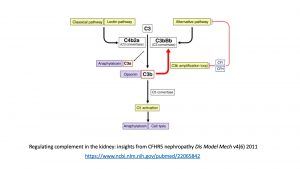
C3 glomerulonephritis is due to a dysregulation in the alternative complement pathway. https://www.ncbi.nlm.nih.gov/pubmed/23125424
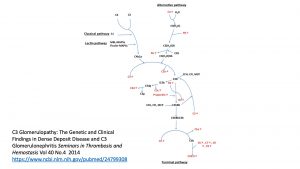
Tuesday
Diagnosis
Patients typically present with hematuria and/or proteinuria in the face of persistently low serum levels of C3. The complement levels in C3 glomerulonephritis unlike infection do not usually resolve within 3 months. https://www.ncbi.nlm.nih.gov/pubmed/27008643
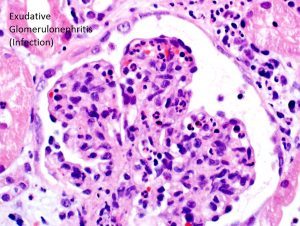
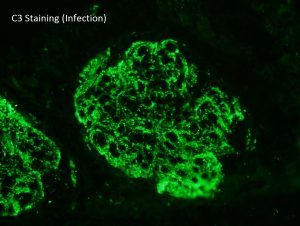
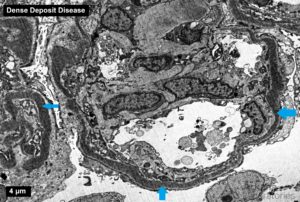
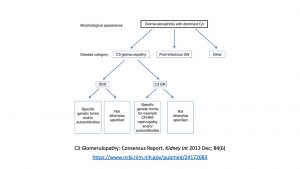
C3 glomerulonephritis is defined as a glomerular lesion which has dominant staining with C3 (little staining with others) and electron dense deposits. By definition, there is an absence of the transformation of the glomerular basement membranes seen in dense deposit disease – see accompanying picture. https://www.ncbi.nlm.nih.gov/pubmed/27056062
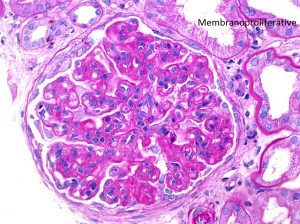
Membranoproliferative pattern is most common in C3 glomerulonephritis
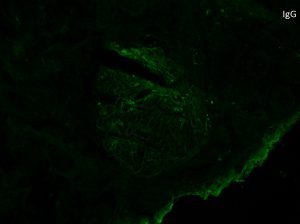
Dominant C3 staining on immunofluorescence is defined as 2 orders of magnitude greater than any other immune reactant
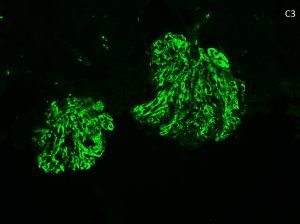
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3842953/
Immunofluorescence should be performed on the paraffin-embedded tissue to look for “masked deposits” which would change the diagnosis. https://www.ncbi.nlm.nih.gov/pubmed/25676556

An underlying infection should be ruled out clinically before labelling a patient with C3 glomerulonephritis

Wednesday
Inhibitors and Mutation
A comprehensive complement panel should be ordered at a specialty laboratory (such as University of Iowa, National Jewish Health Colorado, and Cincinnati Children’s Hospital)
Similar mutations have been reported in dense deposit disease, atypical HUS, and C3 glomerulonephritis.
https://www.ncbi.nlm.nih.gov/pubmed/23125424
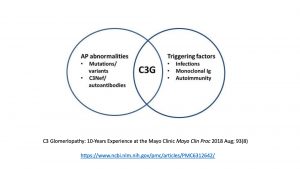
Acquired inhibitors are frequently found in C3 glomerulonephritis patients including autoantibodies to C3 and C5 convertases
https://www.ncbi.nlm.nih.gov/pubmed/24799308
Secondary hits such as infections may unmask a quiescent abnormality within the alternative complement pathway. 33% of patients with C3 glomerulonephritis have an underlying genetic defect. Mutations are found in genes such as CFH, CFHR5, Factor I, and CD46.
https://www.ncbi.nlm.nih.gov/pubmed/29566171
C3 nephritic factor is detected in approximately 50% of cases of C3 glomerulonephritis. C3 nephritic factors are autoantibodies which stabilize C3 convertase.
https://www.ncbi.nlm.nih.gov/pubmed/29670616
Case reports have shown circulating paraproteins can act as an autoantibody to Factor H leading to C3 glomerulonephritis. Any patient over the age of 50 with C3 glomerulonephritis should be checked for a monoclonal protein. https://www.ncbi.nlm.nih.gov/pubmed/23623956
Thursday
Testing, Treatment, Transplantation
Serum levels of complement proteins and their split products, screening for auto-antibodies, and underlying genetic screens included in workup

https://www.ncbi.nlm.nih.gov/pubmed/30692664
Optimal treatment for C3 glomerulonephritis has not been established.
Some patients with C3 glomerulonephritis respond to myophenolate and/or eculizumab. Patients with CFH mutation respond to plasma exchange
https://www.ncbi.nlm.nih.gov/pubmed/23318699
67% of patients develop recurrent C3 glomerulonephritis, 50% of patients lose their graft, and the median time to recurrence is 28 months
https://www.ncbi.nlm.nih.gov/pubmed/24357668
More recent papers have shown long-term favorable outcome for transplant of patient’s with CFHR5 nephropathy despite recurrence of the disease
https://www.ncbi.nlm.nih.gov/pubmed/30844074
Friday
C3 glomerulonephritis is a recently described entity which is due to dysregulation in the alternative complement pathway. Patients typically present with hematuria and/or proteinuria in the face of persistently low serum levels of C3. The annual incidence of biopsy-proven disease is 1 to 2 per million with both sexes affected equally. The median age of diagnosis is 21 years of age, but there is a second spike after the age of 50 due to paraprotein-associated disease. The most common glomerular disease pattern is a membranoproliferative pattern. The hallmark of the disease is dominant C3 staining on immunofluorescence which is defined as two orders of magnitude greater than any other immune reactant. It is prudent to rule out “masked deposits” using immunofluorescence staining on paraffin-embedded tissue and to rule out an infection clinically. By electron microscopy, there are electron dense deposits corresponding to the immunofluorescence staining of C3 (there is no transformation of the GBMs as seen in dense deposit disease). Autoantibodies to C3 nephritic factor are present in approximately 50% of cases of C3 glomerulonephritis. Quiescent abnormalities within the alternative complement pathway are reported in 33% of patients. Optimal treatment for C3 glomerulonephritis has not been established, but some patients respond to mycophenolate and/or eculizumab. 67% of patients who receive a renal transplant have a recurrence of their original disease.
C3 glomerulonephritis has been a success story in precision medicine where the bedside truly has met the laboratory. From the nephrologists who refer patients for specialized complement testing to the basic science laboratory and pharmaceutical industry, there has been shared interest in targeting different steps in the complement pathway. Basic science has found new mutations and drivers of complement, the clinical lab has tested for these mutations, and the pharmaceutical industry has worked on new, targeted therapies. The drug eculizumab (human monoclonal antibody against the complement protein 5) has been given to patients “off label” for C3 glomerulonephritis. Specialty laboratories (University of Iowa, National Jewish Health Colorado, and Cincinnati Children’s Hospital) offer a full complement workup including identification of auto-antibodies, alternative pathway functional assays, and genetic testing of individual complement proteins. Hopefully, over the next few years, there will continue to be a sustained effort to further understand the regulation of the complement cascade.
Athanasiou Y, Voskarides K etc Familial C3 Glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephol 2011 Jun; 6(6):1436-46
Smith RJH, Appel GB, etc C3 Glomulopathy – understanding a rare complement-driven renal disease. Nat Rev Nephrol 2019 Mar; 15(3): 129-143
Quick note: This post is to be used for informational purposes only and does not constitute medical or health advice. Each person should consult their own doctor with respect to matters referenced. Arkana Laboratories assumes no liability for actions taken in reliance upon the information contained herein.